
解读ISO 10993-4:2017《医疗器械生物学评价第4部分:与血液相互作用试验选择》
2018-11-10 08:14侯丽乔春霞赵增琳
中国医疗设备 2018年11期
侯丽,乔春霞,赵增琳
山东省医疗器械产品质量检验中心/山东省医疗器械生物学评价重点实验室,山东 济南 250101
引言
血液相容性评价研究对于与血液接触类的医疗器械非常重要,而国际标准ISO 10993-4:2017《医疗器械生物学评价第4部分:与血液相互作用试验选择》对医疗器械的血液相容性评价有重要的参考意义,因为它给出了评估器械和血液之间相互作用的一般要求和注意事项。本文件由国际标准化组织1992年颁布并于2002年进行了修订(即ISO 10993-4:2002[1])。但是,自2002年以来,该文件只进行了较少修改(即2006年发布的修改单)。由于血液相容性原理和实验方法的复杂性,本文件并未涉及到标准化的评价方法和指标。过去几年来,ISO/TC 194WG9工作组(血液相容性)收到了来自世界各地的数百条意见,经过反复讨论和修改, ISO 10993-4:2017[2]在2017年4月正式批准发布,代表了当前国际上医疗器械血液相容性最新理念和研究进展。本文阐述了最新国际标准的变化,特别是一些当前国内外推荐的医疗器械较常用的血液相容性试验,为国内医疗器械血液相容性评价工作的深入开展提供指导。
1 简化评价流程图
GB/T16886.1《医疗器械生物学评价 第1部分:评价与试验》[3]中规定,对医疗器械新产品进行生物学评价以及上市后医疗器械重新进行生物学评价时,需要对材料进行理化表征以及根据器械是否与同类产品有相同的特性、与人体接触特性和灭菌方法等工艺来确定器械是否需要进行生物学试验。按照生物学评价流程,医疗器械新产品上市前的生物学评价或老产品重新评价后可能不必再进行生物学试验。同样,在ISO 10993-4:2017中进一步强调此理念并简化了评价流程图,特别是与血液直接或间接接触的医疗器械新产品与合法销售的、安全临床使用史的器械相比(即材料、生产过程、几何结构和理化性能、机体接触/临床使用、灭菌工艺/方法/剂量),以确定是否需要进行血液相容性试验(图1)。经过评价后,可能无需进行血液相容性试验而直接得出满足血液相互作用要求的结论,避免了忽视对已有数据的分析和评价及盲目试验、过度依赖试验结果的做法。但是值得注意的是,当器械引入新材料或改变产品工艺方法、增加新颖的器件设计特征、改变临床应用时则可能仍然需要进行血液相容性试验。流程图的修订将血液接触器械的评价策略更加清晰,科学合理。
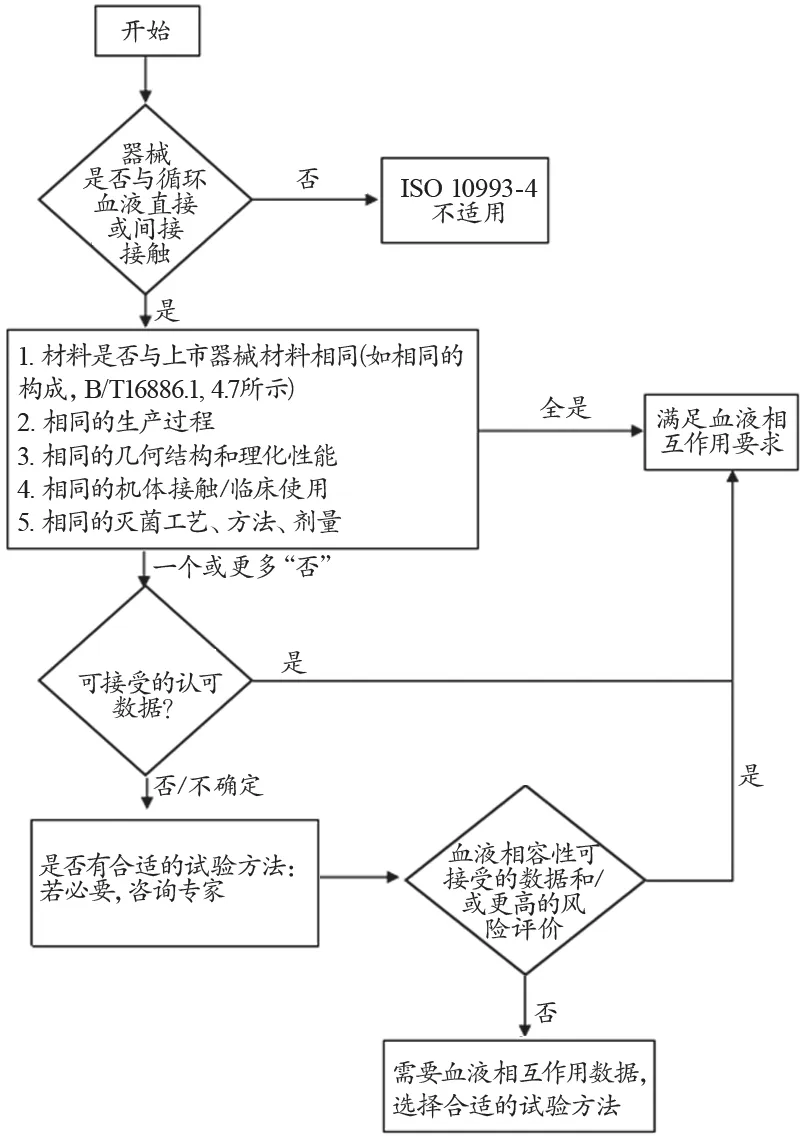
图1 与血液相互作用试验选择判定流程图
2 与血液相互作用试验分类
血栓形成过程主要在体内或半体内条件下发生,在流动或静止的血管或器械区域由凝血、血小板和白细胞激活产生。由于凝血、血小板和白细胞的激活可能会引发血栓形成,ISO 10993-4:2017提出将凝血、血小板、补体和血液学试验作为血栓试验的体外试验,即在某些情况下,凝血、血小板、血液和补体的组合测试可作为血栓形成试验的替代试验。如果进行了体外试验,可能没有必要再做体内或半体内试验(但医疗器械制造商应确认体外试验替代体内试验的适宜性)。由此,本次修订将血液相互作用试验类别由五类(血栓形成、凝血、血小板、血液学、补体系统)改为两大类:溶血试验(材料介导、机械介导)和血栓试验(体外试验和体内/半体内试验)。这也是本版本较重要的修订之处,即将ISO 10993-4:2002中表1和表2合并,并更新了外部接入器械和植入器械的举例和使用试验类别。比如,对外部介入类器械来说,溶血和血液学可能需同时评价而非分开单独评价;对植入类器械来说,按照ISO 10993-4:2017中的要求,只需评价溶血试验(材料介导和机械力介导)和体内或半体内血栓即可(表1)。

表1 与循环血液接触器械或器械部件和适用试验分类——外部接入器械和植入器械
3 血液相容性试验方法
除修订外,一些较少用或不适合医疗器械的试验项目被确定为不常用和不推荐试验的候选。此外,还发展和新增一些新的血液相容性试验方法。随着对血液相容性研究水平的不断提高,新技术和新方法得到完善与发展,尤其是一些商品化试剂盒的开发,积累了丰富的数据,为评估指标的选择提供了良好的基础。本次修订简化了血液相容性试验常用的试验选择,将ISO 10993-4:2002中表3和表4整合成新的表,见表2中血栓试验项目。值得注意的是,将结果与适当的对照(阴性对照)以及相同设计、材料和临床使用的已上市器械/材料的结果进行比较至关重要。
4 血栓试验
当材料与血液接触时,材料表面吸附以蛋白质为主的生物分子,紧接着发生的是同血小板和凝血因子激活为主、补体和白细胞激活为辅的一系列的反应,产生表面血栓和炎性反应等机体反应,故凝血、血小板激活、补体、血液学指标与血栓形成过程密切相关。较上一版相比,ISO 10993-4:2017重要的变化是明确将血栓试验分为体外试验和体内试验,即将凝血、血小板激活、补体、血液学指标作为体外血栓的指标。在很多情况下,如果进行临床相关的体外血栓试验,没有必要再进行体内或半体内试验,这表明将有更多的机会进行有意义的体外试验来替代或补充动物试验。反之,如果进行体内血栓形成试验得到了最终的血栓形成结果,也不需要再评价血小板激活、凝血等血栓起始阶段的指标。

表2 医疗器械与血液相互作用
4.1 体内试验
体内试验与体外试验相比,更能模拟临床最终用途,但由于与血液接触的医疗器械应用的多样性,决定了体内测试模型的多样性,因此选择合适的动物模型、种属差异性、试验成本、动物伦理以及缺乏相应的种属特异性的试剂盒等因素制约了体内试验的进行。对于体内植入的器械进行体内试验时,选择终产品或其部件,应尽可能地模拟临床实际使用条件,如植入部位、几何形状、接触周期、血流情况、无菌操作、抗凝过程等。另外,使用的抗凝剂类型与说明书中的类型和剂量也需要保持一致。
对于要在无抗凝或抗凝条件下静脉环境内植入的导管状器械,特别是评价新导管或新涂层、表征新材料、改变材料供应商、改变工艺的情况时,可选择非抗凝静脉植入模型和抗凝静脉植入模型,即在无或有抗凝剂的情况下,将植入物放置于大型动物(通常为2~3只)静脉至4 h。最常用犬股或颈静脉模型,测试材料/器械位于一侧静脉,对照材料或已上市对照器械放置在对侧部位。为防止器械—器械相互作用和/或血栓形成位置差异带来的偏差,通常采用交替试验和对照的植入位置。测试器械的结果宜等于或小于已上市对照产品的血栓形成。必要时可定制合适的器械样品以保证置入最多15 cm部件长度,同时保证与最终器械相同的材料比例。本版标准还特别指出,植入部位、植入技术、器械—血管壁接触程度、时间/接触期、材料表面状态、取出技术、植入物尺寸相对于血管直径的比例、抗凝剂等因素均对体内血栓试验有较大的影响,但由于器械种类的多样性、实验动物和操作者之间的差异以及当前认知水平的限制,目前还无法对植入模型建立标准化的方法。
试验完成后通过器械表面血栓情况(也可分析观察到的血栓重量和血管通畅性作为补充分析)、末端器官(如肺和肾)的血栓情况以及相关的血液分析进行评价,见(表2)。
4.2 体外试验
正如ISO10993其他部分一样,体外试验是评价医疗器械和材料的生物安全性有效的筛选工具。在进行动物试验和临床研究前,推荐接触血液的医疗器械从早期设计、材料选择及几何形状直到最终成品测试产品开发周期阶段中进行广泛的体外测试。体外试验需要考虑并规范的试验因素为试验系统中全血的体积、血液接触时间、血液温度、血流状况、抗凝剂类型和水平、试验系统本身接触血液的表面积以及与血液接触比例等。试验材料/器械与血液的接触条件越接近临床应用,评价结果的预测性越高。接触后,可根据器械特点选择一个或多个试验类别进行测试,即凝血、血小板、血液学和补体系统等[4]。
4.2.1 凝血试验
凝血途径包括接触激活途径(内源性途径)、组织因子途径(外源性途径)以及内外源凝血的共同凝血途径,其中凝血酶生成并激活了血栓主要成分——纤维蛋白的形成。与血液接触器械/材料相关的凝血主要通过接触激活途径发生,具体反应流程,见图2。

图2 凝血级联反应
凝血产物随着时间的推移具有不同的发展阶段,不同阶段的凝血产物水平可能会出现数量级差异,即当材料/器械与血液混合时,测试样品的影响在每个阶段可能是完全不同的。因此,评价时应在材料/器械与血液接触的活化期进行。
通常采用ELISA试剂盒测定TAT(凝血酶—抗凝血酶复合物)、F1.2(凝血酶原形成凝血酶时释放的蛋白质片段)和FPA(形成纤维蛋白时从纤维蛋白原释放的蛋白质片段)、PTT(ASTM F2382)作为凝血指标(表2)。由于活化物质可掩盖由器械或其材料组分引起的任何活化,因此活化部分凝血活酶时间(APTT)在体外评估血液接触器械/材料的血栓形成时间中不推荐使用。此外,凝血酶原时间(PT)和凝血酶时间(TT)等指标通常也不推荐用于评估接触血液的医疗器械和/或材料。
4.2.2 补体试验
补体是由30余种广泛存在于血清、组织液和细胞膜表面的蛋白质组成。医疗器械与机体相互作用时,补体激活不但可产生免疫毒性、超敏反应和过敏毒素,还可促进和加速医疗器械/材料表面的溶血、血小板、白细胞活化以至于形成血栓。补体激活途径有3种:经典途径、替代途径和甘露糖结合凝集素途径,其中替代途径越过了C1、C4、C2三种补体成分,直接激活C3继而完成C5至C9各成分的连锁反应,是医疗器械最常引起的激活途径。本版标准给出了补体激活替代途径的原理图(图3)。
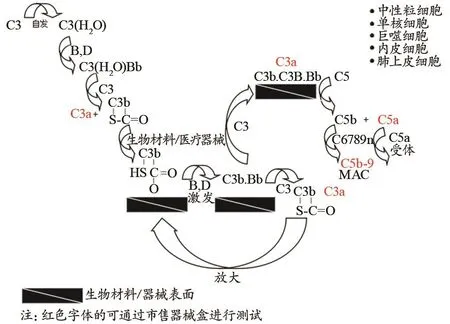
图3 补体激活替代途径的原理图
某些补体成分的升高表明补体系统的激活,目前采用分子生物学方法(主要为ELISA法)检测血液中补体激活的水平。试剂盒的灵敏度是选择补体激活指标的关键因素,如虽然C5a片段被认为是与血液接触器械相关的补体激活中最关键的补体因子之一,但试剂盒对该指标的灵敏度较低,故C5a不是常规的测试指标。经典的补体检测为评估C3a和SC5b-9复合物,其中SC5b-9是补体激活后形成的末端通路产物,通常被认为是代表补体激活程度的较为重要的标记。CH50降低预示总体补体消耗,但CH50在医疗器械血液相互作用试验中一般也不用于评估补体激活。由于补体激活试验是体外试验,方法敏感度较高,但由于不同试剂盒测试参数不同,影响因素较多,实验室间数值差异较大(表2)。
补体激活通常在血液接触器械材料表面后不久的早期阶段发生。体外循环器械特别是与血液大面积接触的器械,如血液透析器和心肺旁路系统等,补体激活是产生其不良反应的重要因素之一。由于补体激活严格依赖表面接触,器械或材料的表面积对补体激活结果影响很大[5],样品制备时注意尽量不要选择浸提液法,而是选择直接接触法,并尽可能提高器械或材料与血液接触的表面积比例,以增加测试的反应灵敏度。但目前尚未得到与临床不良事件相关联的补体激活的器械与血液接触的表面积阈值。
另外,作为有两个相同来源的生物活性系统,凝血和补体之间相互作用存在一定的联系,其结果有一定的相关性。
4.2.3 血小板
血小板与血栓形成有密切的关系,可通过粘附并释放因子和/或聚集成止血栓子促进血栓形成过程。对与血液接触的医疗器械和材料,ISO10993-4:2017推荐的常用指标为血小板计数和血小板脱颗粒(表2)。βTG、PF4是存储在血小板α颗粒中并在血小板激活后大量释放的蛋白质,TxB2是与物种无关的血小板激活标志物,是活化的血小板产生的有效的血小板激活剂。一般可采用细胞分类计数仪测定血小板计数,酶联免疫吸附(ELISA)测定血小板脱颗粒。
血小板激活是一个动态过程,如果从试验系统中取出试验材料或器械后不能立即评价血小板,可添加柠檬酸—葡萄糖、柠檬酸盐—茶碱—腺苷—双嘧啶醇等血小板稳定剂阻止进一步的血小板活化并稳定血小板。另外,凝血级联反应产生的凝血酶是一种有效的血小板激活剂,可以容易地引起血小板脱颗粒。因此,凝血和血小板激活之间存在一定的联系。由于某些物质通过血小板活化而引起血栓形成,而其他物质通过凝血激活而产生血栓,因此有必要对两者同时进行测试。
4.2.4 血液学
血液学评价包含血细胞和血浆成分定量测定(表2)。常用的血液学评价方法为全血细胞计数和白细胞激活。全血细胞计数主要测定血液中白细胞和红细胞的数量或比例以及血小板计数,为器械/材料与血液成分相互作用提供基本信息。通过比较血小板和白细胞在与材料/器械接触前后的计数可推断激活的血小板和白细胞在血栓形成表面数量降低,为器械表面血栓形成提供依据。
4.3 溶血试验
溶血试验不参与血栓形成。一直以来,溶血被认为是一种重要的反映材料与血液相互作用的筛选试验。根据医疗器械的特性和使用方式,引起溶血反应的主要原因有以下三种:渗透压介导的溶血、机械介导的溶血、材料介导的溶血。特别要注意的是除了器械表面接触(直接法)及材料提取物产生的溶血(浸提液法)外,一些器械(如血泵、机械心脏瓣膜、血液过滤器、单采和细胞分离系统等)在机械操作或在流经复杂管路时的流体力学、结构等因素产生的潜在的红细胞变形等破坏作用[6-10]。故除了材料介导的溶血反应外,还应结合临床使用条件,在临床血液动力学条件下(血细胞比容、流速、压力、接触时间等)建立标准化的方法,评价此类型产品在机械力条件下可能产生的溶血潜能。这种标准化的方法应能预测在预期临床使用过程的最差情况下(如最高血流速率等)产生的溶血。目前国际上推荐的标准化方法大多还未涉及到特定产品的方法。
国际上常见的材料介导的溶血试验方法主要有下面几种 :ASTM F756-13[11]、MHLW 法[12]和 NIH 法[13]( 由 于纳米材料的特殊性能,常规试验方法不一定适合纳米材料,故由ASTM F756-13修改后采用的ASTM E2524-08[14]可用于纳米材料的溶血测定)。ISO/TC194WG9工作组自2011年来,采用不同程度的溶血材料、不同浸提条件、不同种属(人/兔)的血源、不同接触方式(直接接触/浸提液)发起了包含山东省医疗器械产品质量检测中心在内的来自5个国家11个实验室的Round Robin试验。经过试验验证,三个试验方法对材料溶血反应的敏感度不同,结果(如NIH法的直接接触法)出现一定的差异[15]。SAC/TC248(医疗器械生物学评价技委会)在2016年起草并报批了行业标准《医疗器械溶血试验 第1部分:材料介导的溶血试验》,可作为我国企业和检验实验室进行溶血试验的方法学参考。
5 应注意的问题
5.1 标准化的试验方法
除了溶血试验外,ISO/TC194工作组近年来已经完成或正在推动的实验室间血液相容性方法比对试验还包括血栓形成体外替代试验、补体激活试验等,但由于器械与血液相互作用过程的复杂性,此次修订的标准仍未列出具体的试验方法。预计在不久的将来,ISO/TC194将制定出医疗器械适用的血液相互作用的试验方法以TR(技术报告)的形式发布,供使用者根据器械临床应用情况,选择最适宜的试验条件并尽可能地使用标准化的方法。
5.2 接触表面积对测试结果的影响
测试器械与血液接触时,凝血、血小板、补体等因素被激活的程度通常与血液接触的器械表面积成比例[16]。由于器械或材料的表面积对测试结果影响很大,在测试时尽可能提高器械或材料的表面积与血液的接触比例以增加测试的反应灵敏度。在每个研究中确认测试是很重要的。为充分评价材料的血液相容性的程度,可以设置表面积/血容量比(接触比)为变量。按照GB/T16886.12《医疗器械生物学评价 第12部分:样品制备与参照样品》[16],可以考虑3.0~6.0 cm2/mL血液(基于器械厚度)的接触比至这个比值的1.5倍和2.0倍以增加血液成分对测试材料的敏感性。
5.3 应设立合理的对照物
由于一些临床可接受的指标水平仍没有确定的通过/失败的阈值,故根据血液相容性试验的特性,试验时同时使用阳性对照、阴性对照或参考材料和已上市同类器械等参照物。值得注意的是,目前并没有一类公认的阳性对照品或材料可在不同指标或试验体系中获得明显的阳性结果,这就需要实验室首先应确认阳性对照物以确保试验体系的有效性和合理性。这样,测量指标的临床意义和由于献血者遗传学和实验室方法等因素造成的结果变异性,可用统计学方法(如方差分析或均值比较)客观地评估和解释。
综上所述,ISO 10993-4:2017在基于ISO 10993-1定义的预期用途和接触时间对血液接触类医疗器械进一步分类,对评价器械与血液相互作用的各种指标、相关的基本原理以及根据预期用途和血液接触持续时间需要考虑的试验选择等内容进行了优化和补充。在医疗器械或材料基础研究和早期开发领域,ISO 10993-4:2017标准的实施可以为确定医疗器械或材料及表面改性或新型医疗器械的血液相容性性能的试验类型、适宜的方法提供进一步的参考指南。
猜你喜欢
护理与康复(2022年6期)2022-11-25
现代仪器与医疗(2022年1期)2022-04-19
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年2期)2021-03-29
河南医学研究(2021年4期)2021-03-10
生物医学工程学进展(2021年3期)2021-01-20
甘肃教育(2020年18期)2020-10-28
质量安全与检验检测(2019年3期)2019-07-31
质量安全与检验检测(2018年6期)2018-12-28
中国医药指南(2017年3期)2017-11-13
