
分子印迹法-高效液相色谱-串联质谱法检测牛奶中的氨基糖苷类抗生素
2018-12-05 13:14李云辉
现代食品 2018年19期
◎ 李云辉
(昭通市质量技术监督综合检测中心,云南 昭通 657000)
通常,牛奶的质量问题在于其中的抗生素残留以及其他一些人为添加的物质等。即使可以控制人为添加物质的方式来提高牛奶质量,但抗生素的残留仍无法被有效控制。氨基糖苷类化合物(Aminoglycosides,AGs)是由氨基环醇、氨基糖以及其他糖类组成的抗生素的总称。它是一种由氨基糖与氨基环醇以过氧桥方式连接而形成的苷类。根据氨基糖苷类的结构特点,可分为5种对环己醇衍生物的分子结构与取代基,且其在实际临床大多是链霉胺衍生物和2-脱氧链霉胺衍生物。这类抗生素在治疗和预防畜牧类动物的疾病方面有十分显著的作用,是牛奶中一大类残留的抗生素来源,其抗菌机制通过阻碍细菌蛋白质的合成来完成,对多种革兰氏阳性菌和革兰氏阴性菌有显著的抗菌效果。长期摄入含有此类抗生素的牛奶会在人体内产生富集作用,副作用很大,有可能导致耳毒性以及肾毒性等疾病。
目前的检测标准方法中牛奶抗生素的检测多是利用微生物抗体反应的定性检测方法,没有对这类化合物直接进行专一检测和定量。目前,国内外学者对氨基糖苷类抗生素的研究大多集中于种类、结构和在原药品中的含量检测,少量有在食品中残留进行检测分析研究报道,但前处理都采用离子交换反向吸附固相萃取柱,或者使用亲水-亲脂平衡固相萃取柱来对待测样品进行净化,通过以多空二乙烯基苯为框架结构,虽具备很好的亲水性和亲脂性和更广泛的pH值适应性。但是,这两类方法依旧无法达到很好的效果,选择性不高,分离效果不佳。
本文提出一种利用分子印迹技术发展而来的固相萃取柱法,以链霉素为模版的方式来实现从复杂基质中对AGs的抗生素进行有效的选择性分离。以期通过低成本、易操作的技术来实现对氨基糖苷类检测前处理,再利用高效液相色谱-串联质谱法[1](HPLC-MS/MS)技术对AGs抗生素在牛奶中的残留量进行检测,达到满意的效果。HPLC-MS/MS通常可以检测牛奶中的β-内酰胺类[2],四环素类[3],大环内酯类[4],磺胺类以及氨基糖苷类。对于利用HPLC-MS/MS来对AGs残留量进行检测的方法,其前处理要求较高,乳制品样品的基质复杂,含有大量的内源性化合物会对分析结果产生一定的干扰。
2 实验中应用到的技术
2.1 HPLC-MS/MS技术的前期处理
HPLC-MS/MS技术成为了现代不可替代的检测分析技术。HPLC是一种高压高效液相分离技术,而质谱的操作需要在真空环境中才能进行;并且在检测过程中液相色谱通常采用无机盐,质谱的缓冲剂一般是采用挥发性的缓冲盐。为了弥补两者使用的一些基本物质上的冲突,HPLC-MS/MS技术中采用了大气压离子化技术来弥补这一缺陷。
2.2 分子印迹技术
分子印迹技术是一种模板分子技术,即通过分子印迹聚合物和聚合物单体接触会形成一个多点的作用,并通过这种聚合过程将记忆的作用下降。在模板分子移除后,聚合物会以模板分子空间结构形式来匹配的多点的孔,这些孔将模板分子及其类似物与识别特性的选择。
其基本步骤如下:①印迹聚合单体的产生。向致孔剂(一种含有模板印迹分子的溶剂)中加入功能单体(常用的有硅氧烷功能单体、偶氮功能单体和复合功能单体),根据共价或非共价作用,使模板分子与功能单体依靠官能团之间产生配合作用,先形成主客印迹聚合单体。②刚性聚合物的形成。加入交联剂、反应剂,在引发剂、光或热等条件下引发单体聚合,使主客印迹聚合单体与交联剂通过自由基产生共聚合,在模板分子周围包裹成高联的刚性聚合物。③洗脱。将刚性聚合物中的印迹分子通过固相萃取柱洗脱和解离出来。
本文通过使用分子印迹技术进行固相萃取,其分子印迹材料的形成过程如图1所示。通过分子印迹技术进行固相萃取,可以对目标痕量分析物产生明显效果的富集作用,即使在基质复杂的情况下也能获得极佳的选择性,同时有效分离和模板分子物化性质相似的待测物质,并且此分析方法的表现也具有很高的稳定性。
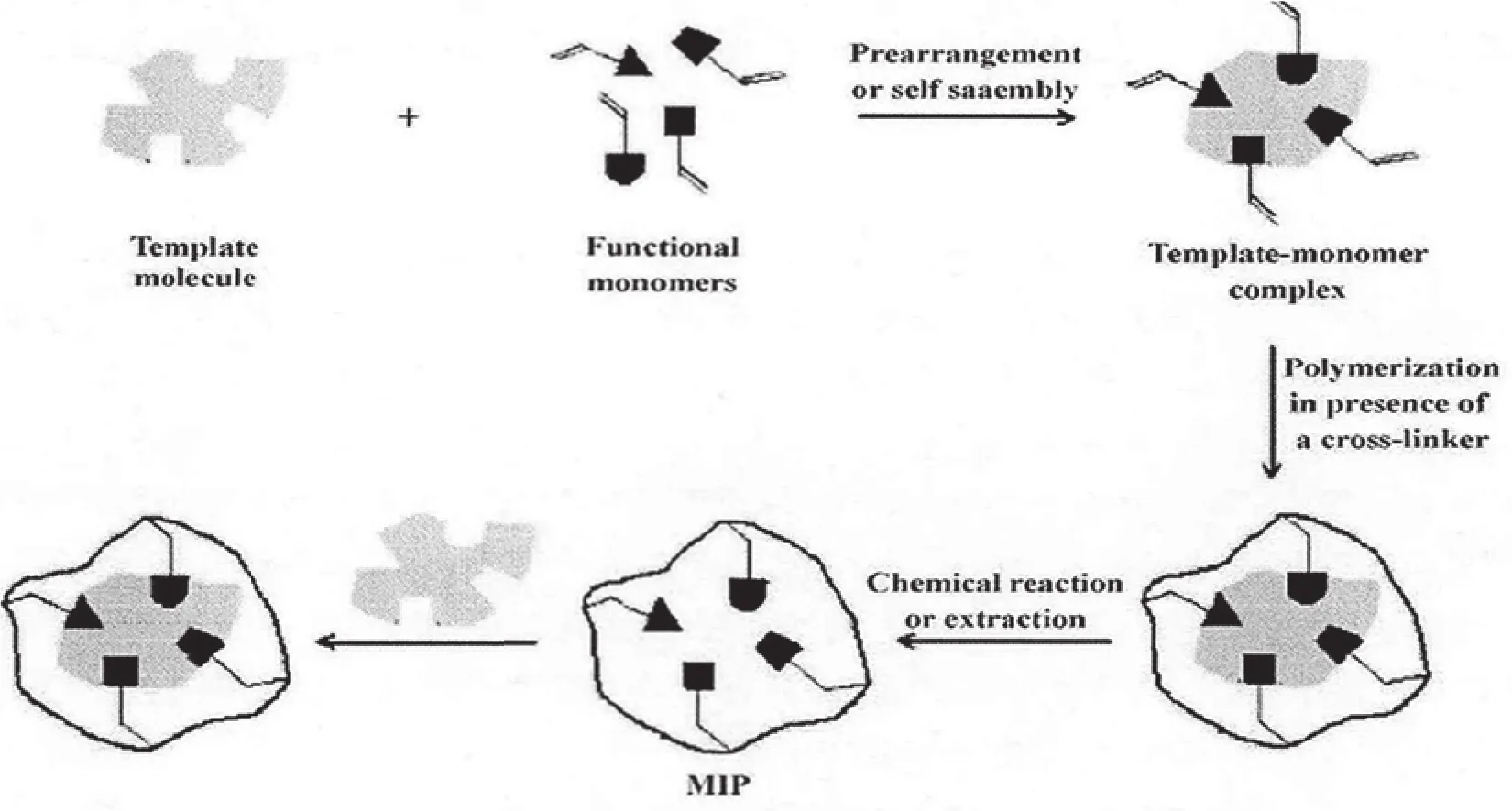
图1 分子印迹材料的形成基本过程图
本文主要以链霉素、双氢链霉素、庆大霉素、卡那霉素、阿普拉霉素和壮观霉素6种AGs药物作为研究对象,基于分子印迹固相萃取柱,高效液相色谱-串联质谱是AGs的检测的一种有效方法。部分AGs的分子结构如图2所示。
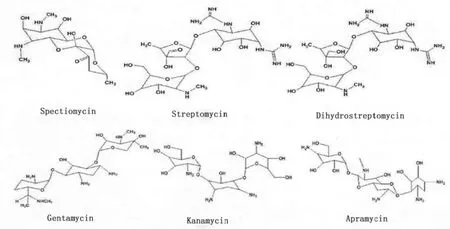
图2 部分AGs的分子结构示意图
3 实验内容
3.1 仪器与试剂
1261-6420A高效液相色谱仪(美国安捷伦公司);AB Sciex 5500 QTrap质谱仪(美国Applied Biosystem公司);Analyst 1.5.1数据釆集及处理系统。壮观霉素、链霉素、双氢链霉素、庆大霉素、卡那霉素、阿米卡星(纯度大于95%),均采购自Dr. Ehrenstorfer公司(德国);甲酸(FA)、甲醇(MeOH)和乙腈(ACN)购自Fisher公司(美国);三氯乙酸(TCA)购自北京检测试剂限公司(中国);Oasis HLB和WCX固相萃取柱是用自Waters公司(美国);实验室用超纯水由MiUipore超纯水机制备。0.1 mol/L氢氧化钠溶液,50%甲酸-甲醇:将甲酸与甲醇以1∶1体积比混合均匀至l00 mL。50%甲酸-乙腈:将甲酸与甲醇以1∶1体积比混合均匀至l00 mL。
3.2 标准溶液的配制
取一定量标准品配制成l 000 mg/L的标准贮备液,用超纯水稀释成l00 mg/L的混合溶液。然后再将此将混合溶液用超纯水进行制成10~5 000μg/L的系列实验溶液。空白基质溶液:按需量取适量溶液,用空白基质配制成50~5 000 μg/L的空白基质溶液,待净化。
3.3 目标化合物提取
量取牛奶约5 mL(精确至0.1 mL)放入50 mL具塞离心管中,向其中再加入4 mL的去离子水并使用机器将其进行均匀的混合。加入20 mL乙腈-水(3∶1,V/V)溶液进行高速振荡2 min后,2 000 r/min再离心8 min,提取上层清夜至另外一支新的鸡心瓶中。用10 mL上述溶液重复提取3次,合并上清液于同一鸡心瓶中。最后使用0.1 mol/L NaOH将溶液的pH值调为8.5,待净化。
3.4 样品净化
将提取液在45 ℃环境下旋转蒸发至余7 mL左右,并加入2 mL 0.1 mol/L磷酸盐缓冲液,将溶液全部上样,再利用固相萃取柱进行处理(使用前依次用3 mL甲醇、3 mL水和3 mL 0.1 mol/L磷酸盐缓冲溶液条件化),然后用3 mL磷酸盐缓冲液冲洗鸡心瓶2次,洗液也移至柱上,用2 mL的初始流动相定容,过滤,待供HPLC-MS/MS分析。
3.5 液相色谱条件
载气:高纯氮气;碰撞气:氩气;源温度:120 ℃;脱溶剂气温度:350 ℃;锥孔电压(Cone):18.0 V;毛细管电压(Capillary):3.0 kV;溶剂气体流量:50.0~1 500.0 L/h锥;碰撞电压(Collision):31.0 V;检测模式:电喷雾电离源(ESI)负离子模式,选择离子检测模式(SIR)对质荷比(m/z573)定量、子离子检测(Daughter)定性。色谱柱:ODS柱(ACQUITY UPLC BEH 1.7 μm、2.1×50 mm), 乙 腈 -0.01% 甲酸溶液(31∶69)为流动相;柱温40 ℃;流速0.2 mL/min,进样量:1 μL。色谱柱(150×3 mm,3 μm);进样量:30 mL;流动相为甲醇(A)-0.01 mL/L三氟乙酸(B),洗脱梯度:0 min(5.0% A)→5.0 min(30.0% A)→10.0 min(33.5% A)→12.0 min(65.0% A)→17.5 min (65.0% A)→18.0 min(5.0% A)→25.0 min(5.0% A),共25 min。
3.6 质谱条件
电喷雾电离源,多反应监测(MRM);ESI+;气帘气(CUR)压力25 psi,雾化气压力60 psi(GSI),辅助气体压力为50 psi(GS)、电喷雾电压4.5 kV,去溶剂温度(TEM)为500 ℃,碰撞室出口电压(CXP)为11 V,Q0入口电压(EP)为15 V。所选6种目标化合物的参数见表1。
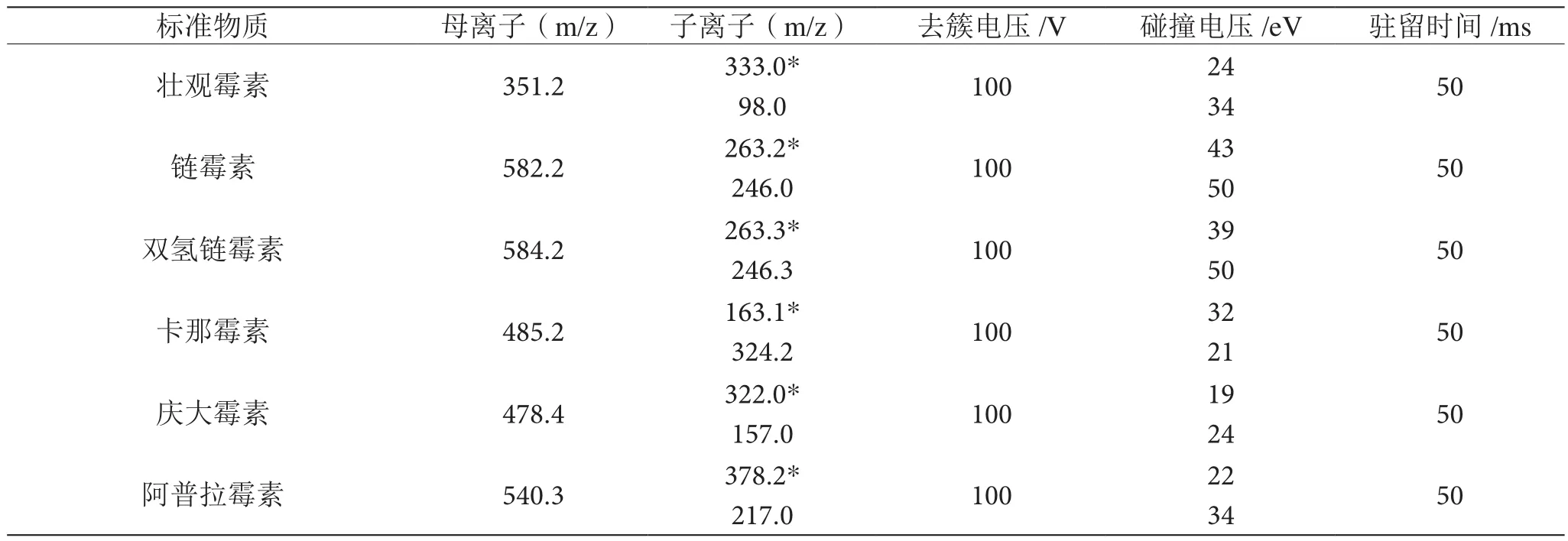
表1 六种目标化合物的参数表
4 结果与讨论
4.1 色谱柱的选择
由于氨基糖苷类物质的极性大,常用的反相柱不能有效地保留。因此,想要增强氨基糖苷类抗生素物质在色谱柱上的停留时间,需在流动相中加入一些易挥发的离子对试剂,如七氯乙酸、三氟乙酸等。而这些离子会残留在仪器中从而影响到仪器的精确度,会增加维护难度。
本文通过HILIC色谱柱分离样品,用流动相缓冲氨基糖苷类抗生素的分离和峰形和响应盐浓度观察HILIC色谱柱。从图3中可知,都使用HILIC色谱柱来分离氨基糖苷类抗生素时,阿普拉霉素和庆大霉素的色谱峰有严重拖尾的现象,影响积分。而链霉素、双氢链霉素、庆大霉素、卡那霉素的实验结果表明,想要获得相对好一些的峰形,可以在流动相中加入较高浓度的缓冲盐(一般为100~140 mmol/L的甲酸铵)。正如前所述,过高的缓冲盐浓度挥发之后会降低响应灵敏度,也会给质谱检测器带来一定的污染。因此,本文采用一种氨基柱作为色谱柱。通过使用氨基柱对6种AGs抗生素的分离结果如图3所示。
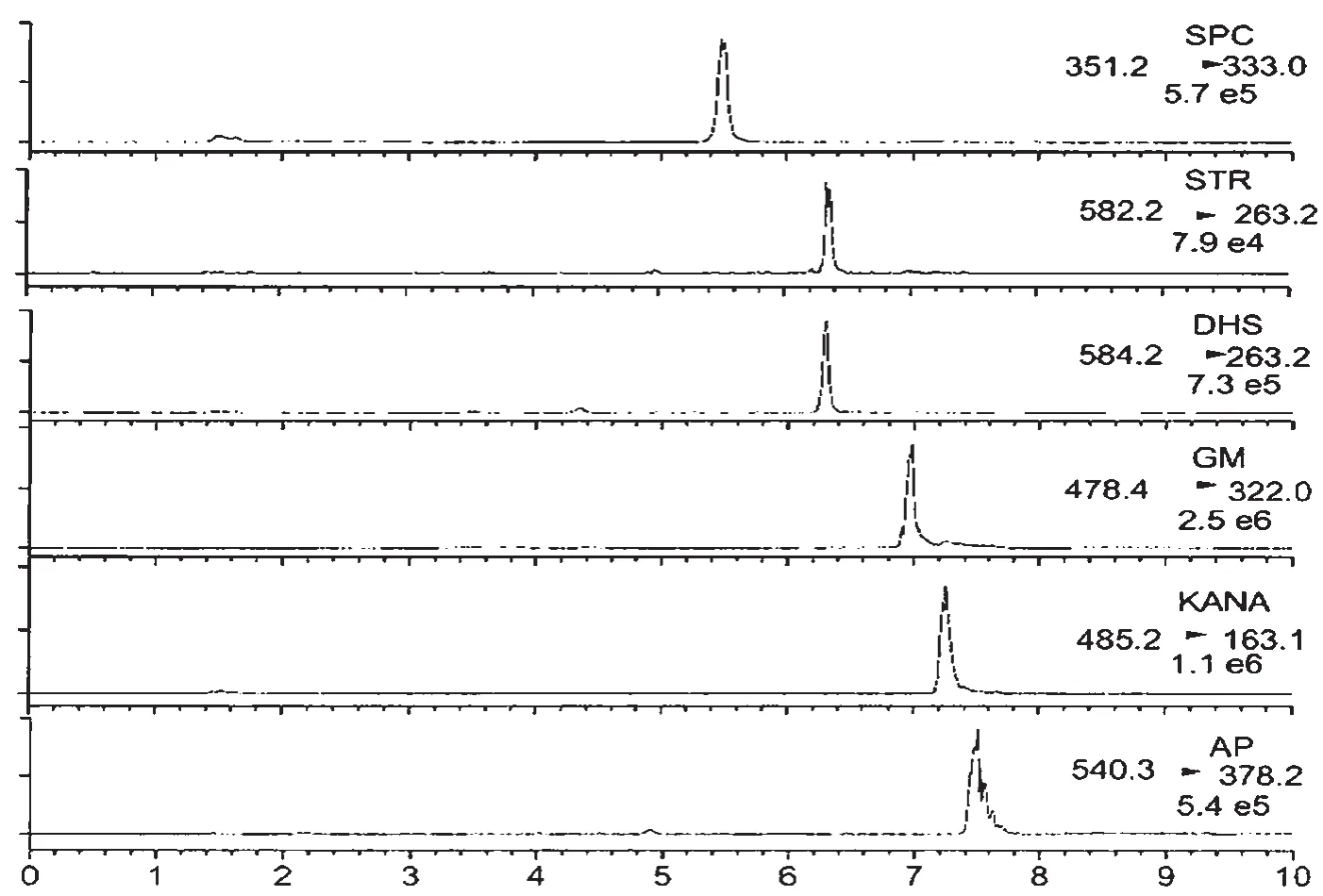
图3 加标样品中提取的离子色谱图
4.2 质谱条件的优化
由氨基糖苷类抗生素的化学结构可知,该类化合物的分子结构中均含有-OH或者-NH2官能团,在质谱上存在十分强的正离子响应,从而通过ESI+模式的扫描方式可以得到很好的分析结果。首先使用1 mg/kg的单标进行针泵进样注射分析,正离子模式下用Qi母离子进行全扫描。在质谱正离子模式下,其一级质谱的氨基糖苷[M+2H]2+或[M+H]+分子离子峰,其规模较大,将分子离子峰选为母离子。之后再对6种AGs的母离子进行二次质谱碎裂,可以得到零散的离子峰的信息。以此推断出待测样品的2个特征离子对,并进一步利用这个信息在多反应监测(MRM)模式下优化去簇电压和碰撞能量等仪器条件,经优化后的6种氨基糖甘类抗生素的质谱参数如表1所示。
4.3 MIP固相萃取柱条件优化
由于氨基糖苷类物质的结构中含有-0H或者-NH2,易与H+相结合,因此,上样液的pH值会对待测样品在固相萃取柱上的保留造成一定程度的影响。本研究中,考察了不同pH值对待测样品保留的影响。使用1 mg/kg的混合氨基糖甘类溶液上样,并提取上样液,进行测试。实验结果表明,当上样液的pH值为7~7.5时,所有待测样品均无穿透。洗脱溶剂的优化考察了浓度为0%、10%、20%、50%,60%、80%甲酸乙腈作为洗脱溶剂时对待测样品回收率的影响。使用1 mg/kg的氨基糖苷类溶液上样,提取洗脱液再进行测试。结果表明,当洗脱溶剂中的甲酸乙腈含量为50%时,6种待测样品的回收率比较可观。
4.4 方法回收率与精密度
在空白基质中添加3个浓度(50、200、1 000 μg/L)的混合标准溶液,对不同添加浓度标液均做了6次平行测试,得到的结果如表2。从表2中可得,对于卡那霉素、庆大霉素和阿普拉霉素3种AGs而言,利用分子印迹技术的方法其回收率达到50%以上,其精密度(RSD)不到10%。对于链霉素和双氢链霉素而言,本方法的回收率分别是30%和40%,且精密度(RSD)也不到10%。对于壮观霉素SPC,本方法的回收率较低仅为5%~9.5%,而且精密度(RSD)为10%~15%。
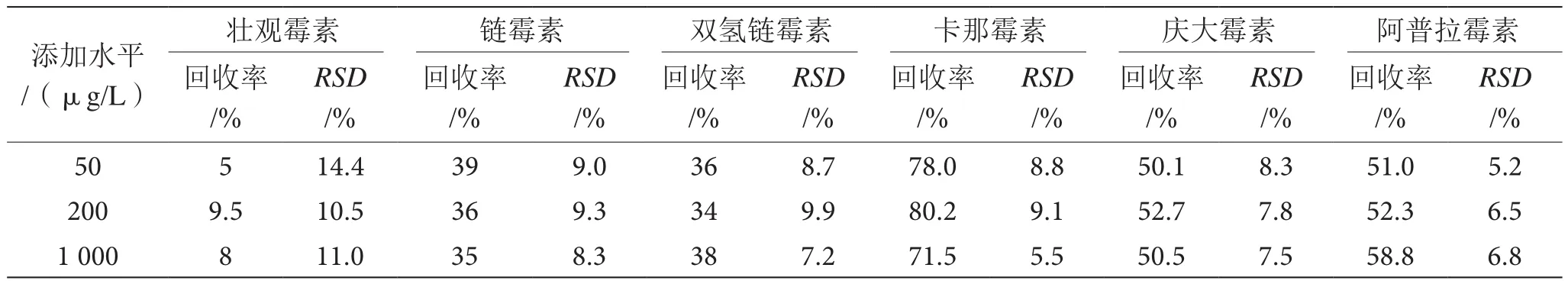
表2 方法回收率与精密度结果表
4.5 实际样品检测
利用此文中分子印迹技术的方法对本实验室于2016年5—12月期间的100个乳制品样品进行AGs药物的检测,未检出AGs。
5 结论
本研究设计的分子印迹固相萃取小柱,能针对牛奶中氨基糖苷类抗生素实现快速、准确、灵敏检测高效液相色谱-串联质谱方法。考察了不同填料的固相萃取小柱对于牛奶中6种氨基糖苷类抗生素的净化效果,相比于商品化的固相萃取柱,自制的固相萃取可以有效地净化乳基,降低基体效应,提高反应的灵敏度。然而,由于样品前处理阶段的渗透现象仍然存在,希望能够深入下一步的研究。
猜你喜欢
陶瓷研究(2022年3期)2022-08-19
中草药(2022年9期)2022-05-06
云南画报(2021年10期)2021-11-24
化工自动化及仪表(2021年3期)2021-06-04
食品工程(2020年4期)2021-01-20
中国油脂(2020年3期)2020-04-10
酿酒科技(2019年10期)2019-11-12
小学生优秀作文(高年级)(2018年4期)2018-09-11
天然产物研究与开发(2018年8期)2018-09-10
中国马铃薯(2015年5期)2016-01-09
