
头孢氨苄水解物的镍/锌单核配合物的合成、晶体结构及抗肿瘤活性
2018-12-10 06:49王国平
无机化学学报 2018年12期
杨 晨 王国平
(浙江大学化学系,杭州 310027)
0 引 言
头孢菌素是一类重要的抗感染药物,具有抗菌作用强、耐水解、高效、毒性小等优点,是目前临床应用最广的抗生素。头孢氨苄作为目前临床使用量较大的一种半合成、广谱头孢菌素[1],能通过抑制细菌细胞壁的合成,对革兰氏阳性菌和革兰氏阴性菌均表现出很强的抗菌活性。研究发现,许多药物以金属配合物的形式存在时拥有较好的可修饰的毒理学和药理学特性[2],甚至有明显的抗肿瘤活性[3-6]。头孢氨苄含有丰富的潜在供体原子,如侧链氨基、羧基、β-内酰胺和酰胺基团,可有效地作为配位剂与金属离子反应形成稳定的配合物[7]。近年来对头孢菌素金属配合物的配位行为和抗菌活性的研究较为广泛,发现部分头孢菌素与金属配位后其抗菌活性可明显增强[8-13],但是对其抗肿瘤活性研究甚少。最近研究发现头孢吡肟与金属锰反应形成配合物后可以有效抑制人类乳腺癌细胞的增殖并诱导其凋亡[14],这说明该类配合物是一种潜在的细胞凋亡诱导物,可在未来应用于癌症治疗。
因此,本文以头孢氨苄为原料,选用镍和锌金属离子在弱酸性条件下合成2个单核配合物[Ni(cepha)2]·6H2O (1)和[Zn(cepha)2]·6H2O (2)(cepha=cephalosporoate)。使用X射线单晶衍射分析确定其晶体结构,并通过元素分析、红外光谱、热重分析和X射线粉末衍射分析对其进行表征。在此基础上通过MTT法研究配合物对人类肝癌细胞 (HepG-2)和人类乳腺癌细胞(MCF-7)的抗肿瘤活性。
1 实验部分
1.1 试剂和仪器
所用试剂均为市售分析纯,头孢氨苄购于Sigma且使用前未进一步纯化。红外光谱采用KBr压片法,在Nicolet FT-IR型红外光谱仪上测试,测定范围为4 000~400 cm-1;元素分析采用 Perkin-Elmer 1110元素分析仪测定配合物 C,H,N含量;热重分析采用 MATTLER TGA/DSC1 1100仪,升温速率为10℃·min-1,升温至800℃;X射线粉末衍射采用 Ultima Ⅳ(Cu Kα 射线,λ=0.154 056 nm)测定,工作电压为40 kV,工作电流为30 mA,X射线的入射角为 1°,在 5°~50°的 2θ角度范围内连续扫描,扫描速度为 10°·min-1。
1.2 配合物的合成
1.2.1 [Ni(cepha)2]·6H2O (1)的合成
准确称取 0.074 g(0.2 mmol)头孢氨苄和 0.025 g(0.1 mmol)醋酸镍,分别溶解于10 mL蒸馏水和5 mL甲醇溶液,并在搅拌状态下将头孢氨苄溶液逐滴加入到乙酸镍溶液中得到混合溶液。将该混合溶液过滤,滤液置于室温下挥发溶剂。大约2周后,得到蓝色的针状晶体。产率:50.5%。元素分析按C32H48N6NiO16S2的计算值(%):C 42.87,H 5.36,N 9.37;实验值(%):C 42.18,H 5.50,N 9.46。
1.2.2 [Zn(cepha)2]·6H2O (2)的合成
准确称取 0.148 g(0.4 mmol)头孢氨苄和 0.044 g(0.2 mmol)醋酸锌,分别溶解于10 mL蒸馏水和10 mL甲醇溶液,并在搅拌状态下将头孢氨苄溶液逐滴加入到乙酸锌溶液中得到混合溶液。将该混合溶液过滤,滤液置于室温下挥发溶剂。大约4周后,得到黄色的针状晶体。产率:49.8%。元素分析按C32H48N6ZnO16S2的计算值(%):C 42.55,H 5.32,N 9.31;理论值(%):C 42.09,H 5.43,N 9.30。

Scheme 1 Syntheses of complexes 1 and 2
1.3 单晶结构测定
选择大小合适的单晶,粘在玻璃细纤维上,采用Oxford Gemini A Ultra型X射线单晶衍射仪,在293(2)K 用经过石墨单色器的 Mo Kα 射线(λ=0.071 073 nm)作为入射光源,采用ω-2θ扫描方式收集衍射点数据,并对衍射数据进行了线性校正、经验吸收校正、Lp校正。晶体结构解析采用SHELXS-2016程序中的直接法解出[15],其它非氢原子采用全矩阵最小二乘法和各项异性进行修正。氢原子由Fourier法找出或用理论加氢。所有原子的散射因子均来源于国际晶体学表。配合物1和2的晶体学和精修参数见表1。
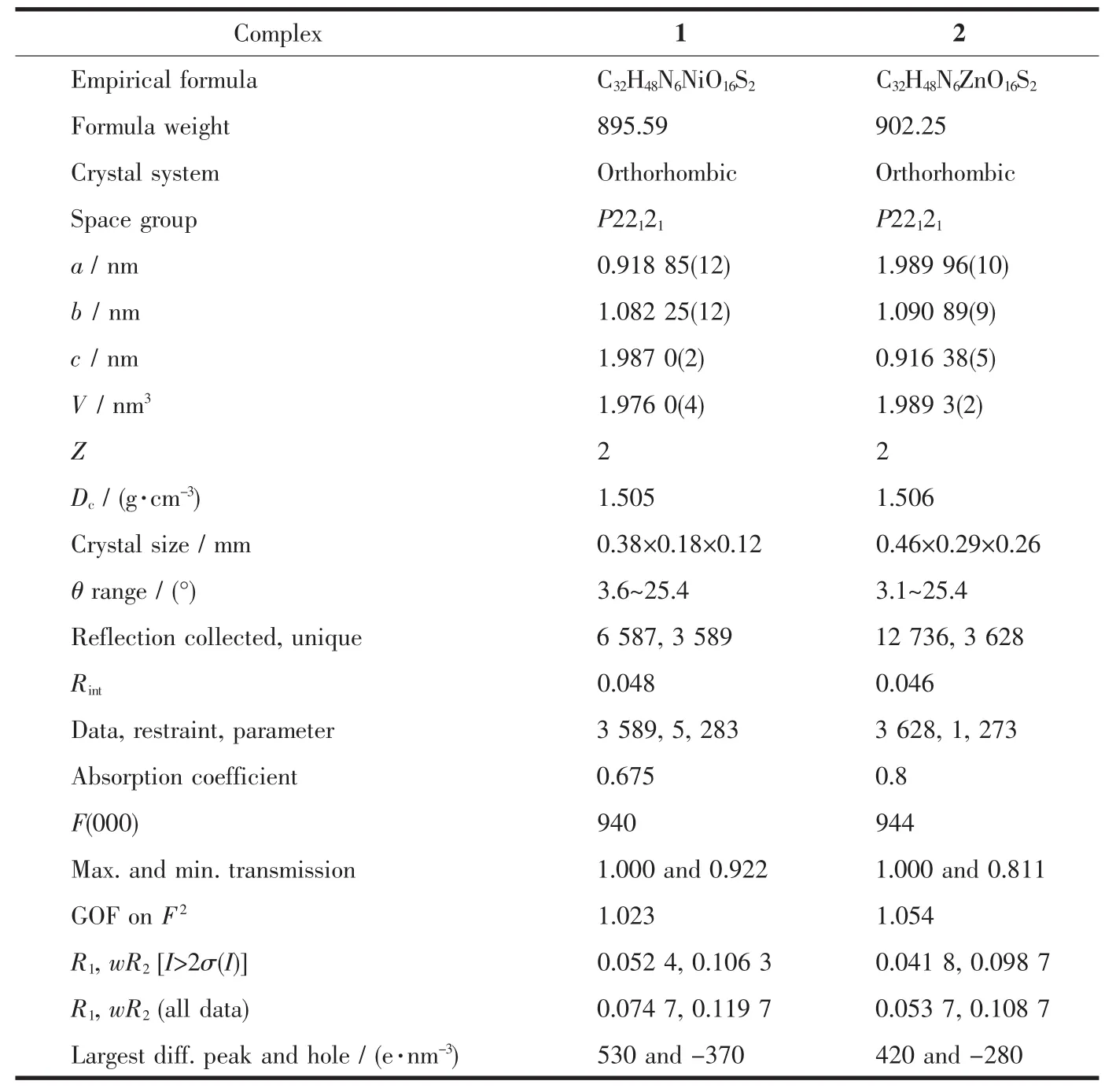
表1 配合物1和2的晶体学和精修参数Table 1 Crystal data and structure refinement for complexes 1 and 2
1.4 体外抗肿瘤活性测定
采用MTT比色法测定醋酸镍、醋酸锌、配体及配合物 1、2 对 MCF-7 (人类乳腺癌细胞)(24 h)和HepG-2(人类肝癌细胞)(12 h)的抑制增殖活性。测试方法同文献[16]。
2 结果与讨论
2.1 晶体结构
配合物1和2的晶体结构见图1,部分重要的键长和键角以及氢键参数分别在表2和表3中列出。
配合物1为单核镍配合物,属于正交晶系,P22121空间群。中心镍ギ离子的配位数为6,分别与来自2个配体分子的N原子和4个O原子配位。配位 键 Ni1-O1,Ni1-N1,Ni1-O3 的 键 长 均 不 等 , 在0.204 9(4)~0.206 2(4)nm 范围内。配位键角 O1-Ni1-O1i,O1-Ni1-O3,O3-Ni1-O3i,O3-Ni1-O1i在 87.2(2)°~92.56(16)°范围内,接近 90°,即 O1,O1i,O3,O3i基本处于同一平面。 配位键角 N1-Ni1-N1i为 166.1(3)°。因此,中心镍ギ离子形成以 O1,O1i,O3,O3i作为赤道平面,N1,N1i占据轴向位置的扭曲八面体几何构型。
头孢氨苄在配位过程中发生了较大的变化。由于反应体系为弱酸条件,使得侧链的氨基N原子质子化,且因为金属离子的存在促进了头孢氨苄的水解,β-内酰胺环上的C-N键断裂,形成一个新的羧基[17-18],从而得到了相应的头孢菌素中间体cephalosporoate (cepha)[19]。配体 cepha 上的 O 原子和双氢噻嗪环上的N原子与金属离子配位形成稳定的六元环。邻位的羧基去质子化,其上的O原子与金属离子配位形成一个稳定的五元环。双氢噻嗪环上的N1-C2 和 C2-C3 键长分别为 0.127 8(5)和 0.151 1(8)nm,但在头孢氨苄晶体结构中相应的键长分别为0.141 5(1)和 0.133 3(2)nm[20],对应于 C-N 单键和 C=C双键,说明头孢氨苄的双键位置从3位-4位移动到4位-5位,形成C-C单键和C=N双键,这与头孢氨苄水解的结果一致[21-22]。

图 1 配合物 1 (a)和 2 (b)的 ORTEP 图Fig.1 ORTEPview of complexes 1 (a)and 2 (b)

表2 配合物1和2的部分键长(nm)和键角(°)Table 2 Selected bond lengths(nm)and angles(°)for complexes 1 and 2

表3 配合物1和2的氢键参数Table 3 Structural parameters of hydrogen bonds for complexes 1 and 2

续表3

图2 配合物1的晶胞堆积图Fig.2 Crystal packing diagram of complex 1 from b axis
配合物1的堆积图如图2所示,配合物分子中的配体分子上未配位的羧基O原子,侧链氨基N原子,侧链酰胺羰基O原子以及晶格水分子的存在,使配合物形成大量N-H…O和O-H…O形式的氢键,并依靠这些分子间氢键作用将单核配合物分子组装形成三维网格结构。
配合物2与配合物1结构相同。
为了进一步检验配合物的纯度,我们对配合物1和2进行了X射线粉末衍射分析,如图3所示。通过对比可以发现配合物1和2的特征峰位置与单晶结构模拟的X射线粉末衍射图谱一致,表明配合物为高纯度单晶。

图3 配合物1和2的X射线粉末衍射图Fig.3 PXRD patterns for complexes 1 and 2
2.2 配合物的红外光谱
头孢氨苄及配合物1和2的红外光谱如图4所示。可以看到配合物1和2的红外谱图与头孢氨苄的红外谱图存在明显的差异。头孢氨苄在1 758 cm-1处出现了 β-内酰胺环的 ν(C=O)特征吸收峰,但配合物谱图中该特征吸收峰消失了,表明头孢氨苄的β-内酰胺环参与配位或者发生水解[23]。头孢氨苄的另一个特征吸收峰出现在1 690 cm-1,对应于侧链酰胺的ν(C=O)吸收峰。而配合物的红外光谱中相应吸收峰没有发生较大的位移,表明侧链酰胺没有直接参与配位。同时,配合物在1 600和1 380 cm-1附近出现新的吸收峰,分别对应于羧酸盐的不对称伸缩振动 νas(COO-)和对称伸 缩 振 动 νs(COO-)。 这是羧基参与配位的依据,且1和2的分离值 (Δν=νas(COO-)-νs(COO-)分别为 207 和 218 cm-1,表明羧基以单配位模式与金属配位[24],这与单晶结构分析结果一致。头孢氨苄中的β-内酰胺和双氢噻唑环共有的ν(C-N)在1 354 cm-1出现特征吸收峰,但配合物中该特征峰消失了,表明β-内酰胺和双氢噻唑环上的N原子参与配位[2,25]。此外,与配体相比配合物的红外光谱在600~400 cm-1处发生了很大的变化,在这一区域出现了金属与氧原子成键的特征吸收峰 ν(Ni-O)=504 cm-1和 ν(Zn-O)=507 cm-1以及金属和氮原子成键的特征吸收峰ν(Ni-N)=483 cm-1和ν(Zn-N)=480 cm-1。头孢氨苄的红外光谱还在 3 000~3 200 cm-1范围内出现了一组吸收峰,为氨基的ν(N-H),但配合物中的该谱带保持不变,且在2 600 cm-1处的宽峰可归因于 R-NH3+的 ν(N-H),显示侧链氨基单质子化而没有参与配位。

图4 头孢氨苄、配合物1和2的红外光谱Fig.4 IR spectra of cephalexin,complexes 1 and 2
配合物在3 490 cm-1有一组可归属于水分子ν(O-H)的强而宽吸收峰,说明配合物中可能含有配位水或结晶水,但在 550 cm-1没有出现 ρw(H2O)的特征吸收峰表示水分子并没有参与配位。因此,配合物中的水分子都是结晶水。
2.3 配合物的热重分析
为了研究配合物的稳定性,我们对其进行了热重分析,图5为配合物1和2从室温到800℃的TG曲线。从配合物1的TG曲线可以看出,其分解过程主要分为3个步骤:首先从室温到160℃范围内,出现明显的失重现象,质量损失为12.3%(计算值12.1%),对应于配合物1失去全部6个水分子。随着温度升高,配合物1在一段时间内较为稳定。当温度升至200℃时左右时再次出现失重现象,这主要是无水配合物的进一步分解。该过程较为复杂,可解释为配合物的双氢噻嗪环的一侧发生热分解,质量损失为 47.2%(计算值 47.4%)[26-27]。 最后,在500~800℃范围内,剩下的苯环部分进一步分解,质量损失为30.1%(计算值30.2%)[28]。配合物2的热分解过程与配合物1相似,先失去6个水分子,并且在200℃左右无水配合物进一步分解。配合物1和2热分解最后剩余的固体残渣为相应的金属氧化物[29],对应质量分别为 9.4%(计算值 8.4%)和 9.5%(计算值 9.2%)。
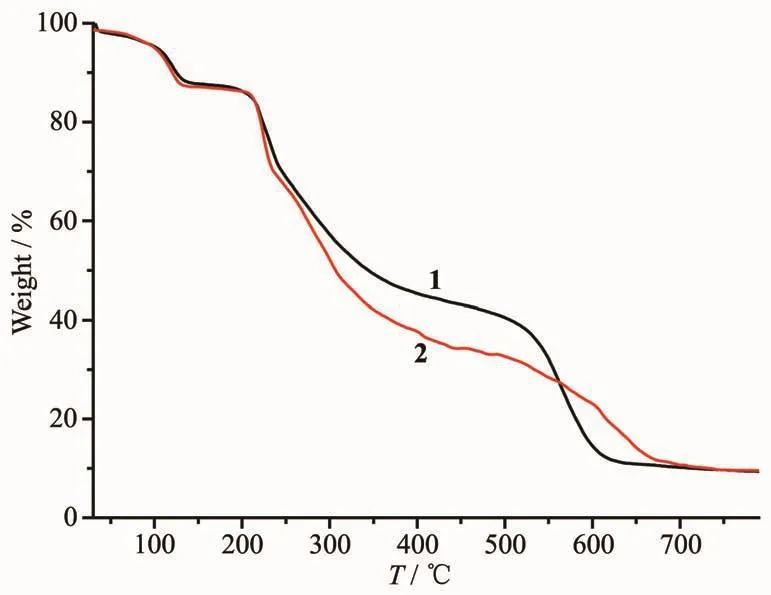
图5 配合物1和2的TG曲线Fig.5 TGcurves of complexes 1 and 2
2.4 配合物的体外抗肿瘤活性
为研究配合物的体外抗肿瘤活性,采用MTT比色法测定了金属醋酸盐、头孢氨苄、配合物1和2对肿瘤细胞抑制增殖活性,结果如图6所示。头孢氨苄单配体随着浓度的增加,对乳腺癌细胞(MCF-7)和人肝癌细胞(HepG-2)的抑制效果均不明显。但是配合物对MCF-7细胞有一定的抑制活性,且随浓度增大而逐渐增强,表明其对肿瘤细胞的抑制活性存在剂量依赖性。而配合物对HepG-2细胞的抑制增殖作用在低浓度时并不明显,当浓度高于100 μmol·L-1时,细胞存活率明显下降。在相同浓度下,配合物2对MCF-7细胞的活性强于配合物1。此外,相应的金属醋酸盐对MCF-7和HepG-2也有一定抑制活性。这说明金属元素的参与,增强了配合物的抗肿瘤活性,且金属元素的种类对配合物的抗肿瘤活性存在影响。

图6 金属醋酸盐、头孢氨苄、配合物1和2在不同浓度下对MCF-7(a)和HepG-2(b)细胞活力影响曲线Fig.6 Cell viability curves of MCF-7 (a)and HepG-2 (b)at different concentration of metal acetates,cephalexin,complexes 1 and 2
3 结 论
以头孢氨苄为原料合成锌和镍的配合物时,头孢氨苄在配位过程中发生水解,产生相应的头孢菌素中间体cepha。配体cepha的氧原子和双氢噻嗪环上的氮原子以及侧链羧基氧原子一起与金属离子配位,得到2个结构类似的单核配合物[Ni(cepha)2]·6H2O (1)和[Zn(cepha)2]·6H2O (2)。1 和 2 均通过分子间氢键将配合物组装成稳定的三维网状结构。2种配合物在200℃时仍有较好的热稳定性。通过MTT法研究配体、醋酸盐和配合物的体外抗肿瘤活性,结果表明头孢氨苄对乳腺癌细胞(MCF-7)和人肝癌细胞(HepG-2)没有抑制增殖作用,但2个配合物对2种癌细胞则表现出一定的体外抗肿瘤活性,且配合物2对MCF-7细胞的抑制活性强于配合物1。
猜你喜欢
山东农业大学学报(自然科学版)(2021年3期)2021-07-29
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
无机化学学报(2020年7期)2020-07-20
世界农药(2019年2期)2019-07-13
当代陕西(2019年6期)2019-04-17
无机化学学报(2018年8期)2018-08-01
粘接(2017年4期)2017-04-25
原子与分子物理学报(2015年3期)2015-11-24
外语学刊(2014年3期)2014-12-03
郑州大学学报(理学版)(2013年2期)2013-03-11
