
表/界面水的扫描探针技术研究进展∗
2019-01-25 09:53尤思凡孙鲁晔郭静裘晓辉江颖
物理学报 2019年1期
尤思凡 孙鲁晔 郭静 裘晓辉 江颖
1)(北京大学物理学院量子材料科学中心,北京 100871)
2)(国家纳米科学中心,北京 100190)
3)(中国科学院大学,北京 100049)
4)(北京师范大学化学学院,北京 100875)
5)(量子物质协同创新中心,北京 100871)
6)(中国科学院大学拓扑量子计算卓越创新中心,北京 100190)
(2018年12月14日收到;2018年12月20日收到修改稿)
表面和界面水在自然界、人们的日常生活以及现代科技中无处不在.它在物理、化学、环境学、材料学、生物学、地质学等诸多基础学科和应用领域起到至关重要的作用.因此,表面和界面水的功能与特性的研究,是水基础科学的一项核心任务.然而,由于水分子之间氢键相互作用的复杂性,及其与水-固界面相互作用的竞争,使得表(界)面水对于局域环境的影响非常敏感,往往需要深入到分子层次研究其微观结构和动力学过程.近年来,新型扫描探针技术的发展使得人们可以在单分子甚至亚分子尺度上对表(界)面水展开细致的实空间研究.本文着重介绍几种代表性的扫描探针技术及其在表(界)面水体系中的应用,包括:超高真空扫描隧道显微术、单分子振动谱技术、电化学扫描隧道显微术和非接触式原子力显微术.此外,本文还将对表(界)面水扫描探针技术研究面临的挑战和未来发展方向进行了展望.
1 引 言
水作为人们赖以生存的资源一直备受关注.火星上水的遗迹给了人们移民外星的可能;南水北调工程则牵涉到数以千万国人的水源.除了这样涉及人类福祉和未来的探索,水也是科学界关注的焦点.分子结构看似简单的水却具有很多独特的物理和化学性质.水的奇特性质与水的微观结构密切相关,尤其是水分子之间的氢键扮演着非常核心的角色.由于氢键相互作用的复杂性,至今水仍然是自然界最为神秘的物质之一.在庆祝《Science》创刊125周年之际,该刊公布了125个最具挑战性的科学问题,其中就包括“水的结构如何?”2015年,《德国应用化学》也将水的相关问题列入未来24个关键化学问题(位列第4).水与外界的相互作用是通过“表面和界面”接触实现的,因此水-固界面是很多物理和化学过程发生的重要场所,研究表(界)面水的微观结构和相关动力学过程对于理解和操控水的特性有着关键的作用.表(界)面水的研究涉及到表面科学、催化、电化学、人工降雨、机翼防冻等从基础到应用诸多领域,并在其中处于核心地位.
表(界)面水不仅涉及水-水相互作用,还涉及水-固体相互作用,这两种相互作用的竞争,决定了表(界)面水的很多独特性质,比如界面水的氢键网络构型、质子转移动力学、水分子分解、受限水的反常输运等.表(界)面水的常规研究实验手段是光谱、核磁共振、X射线晶体衍射、中子散射等谱学和衍射技术.然而,这些研究手段有一个共同的问题,就是空间分辨能力都局限在几百纳米到微米的量级,得到的信息往往是众多水分子叠加在一起之后的平均效应,无法得到单个氢键的本征特性和氢键构型的统计分布.而表面结构通常具有纳米甚至原子尺度的不均匀性,水分子受局域环境的影响会变得尤为明显,这导致对实验结果的分析和归因往往很困难,一般需要结合复杂的理论计算和模拟.因此,非常有必要深入到分子层次上对表(界)面水的微观结构和动力学过程进行高分辨探测,挖掘影响氢键相互作用的物理根源.
近年来,新型扫描探针技术的发展使得人们可以在单分子甚至亚分子尺度上对表(界)面水展开细致的实空间研究,取得了许多重要的进展,大大加深了人们对于表(界)面水的认识.本文着重介绍几种具有代表性的扫描探针技术及其在表(界)面水体系中的应用,包括:超高真空扫描隧道显微术、单分子振动谱技术、电化学扫描隧道显微术和非接触式原子力显微术.此外,本文还对表(界)面水扫描探针技术研究的挑战和未来发展方向进行展望,以期引起更多研究者的兴趣,推动表(界)面水科学和技术的发展.
2 新型扫描探针技术的进展和应用
2.1 超高真空扫描隧道显微术
2.1.1 扫描隧道显微镜简介
1982年,IBM瑞士苏黎世实验室的科学家Binning等[1]发明了扫描隧道显微镜(scanning tunneling microscopy,STM),STM是利用针尖和样品之间的隧道电流对于样品表面进行表征的.由于隧道电流和针尖-样品间距离成指数依赖关系,因此STM对距离极其敏感,具有很高的空间分辨率.图1是STM的工作原理示意图,针尖由一个可以三维移动的压电陶瓷组控制实现针尖在平行于样品表面内的扫描,反馈回路可以测量电流的大小并且控制针尖和样品的距离.这就决定了STM具有恒流和恒高两种工作模式,恒流模式下得到的是表面形貌的图像,恒高模式下得到的是隧穿电流大小的空间分布.两种模式之间是相互关联的,都可以给出样品表面结构的信息.
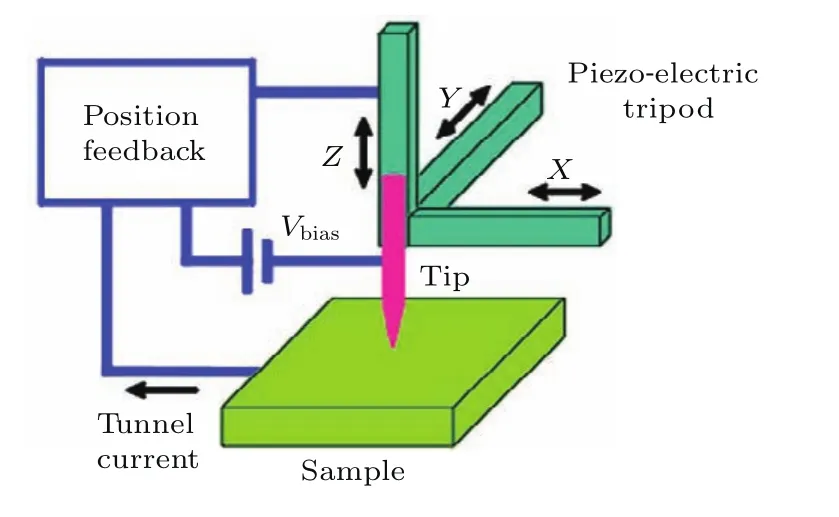
图1 STM工作原理示意图Fig.1.Working principle of an STM.
由于水-水相互作用与水-表面相互作用之间的微妙竞争关系,固体表面的水往往非常复杂,且对于表面的缺陷异常敏感.为了得到精细的氢键网络结构,原子尺度的表征往往更具有说服力.STM就是这样一种精密的仪器,可以对固体表面的吸附分子进行单分子甚至亚分子级别的成像.它主要利用针尖电子与分子轨道之间的隧穿来获得分子的电子结构信息,但是大气环境会使得表面在原子尺度上不断地发生着各种各样的变化,这严重影响了STM的高分辨成像.获得更为“干净”表面的通常手段是在超高真空下进行实验.
2.1.2 超高真空扫描隧道显微术在水-固界面的应用
原子级空间分辨使得STM可以在实空间观察到单个水分子和复杂的氢键网络结构.因为STM实验需要导电的样品,所以过去二十年大部分工作集中在金属表面的水.单个水分子的STM图像通常表现为位于金属原子顶位的圆形突起,并且没有任何内部结构[2,3].主要原因有两个:首先是水的前线轨道是远离费米能级的,其次是水和金属传导电子的杂化可能淹没了分子轨道的信息.水在金属表面的团簇化和浸润已经被一些综述文章很好地总结[4-9].基本的共识是,固体表面的水层吸附并没有普适模型,通常获得的结构是水分子间氢键相互作用和水-金属成键相互作用的精细平衡,因而导致了各种各样的不同于传统“双层冰”模型的氢键网络结构,比如:Cu(110)[11-13],Ag(111)[14,15],Pd(111)[16,17],Pt(111)[18,19],Ru(0001)[16,17,20]以及Ni(111)[21].
近期,金属表面多层水的研究开始受到广泛的关注.但由于水是很好的绝缘体,多层水的高分辨STM研究仅限于少层水(小于三层).Maier等[10]发现在Ru(0001)和Pt(111)表面的第二层六角有序的水层可以诱导第一层无序的水层转变成有序的结构.图2是晶化后的第一层和第二层水分子的原子分辨,可以看到这两层水分子是相互匹配的六角结构,而且水分子都是位于下面衬底的金属原子定位.Lechner等[22]发现继续在Pt(111)表面的单层水上沉积NH3分子会诱导水分子调整方向,直到有一个OH和NH3形成一个氢键,密度泛函理论计算给出了这个过程不存在能量势垒.

图2 Ru(0001)表面水团簇的原子结构图 (a)晶化的两个水层的原子分辨,团簇周围被裸露的钌环绕着,插图中蓝色的点显示了第三层水分子吸附的位置;(b)第一、二层水都和衬底原子对齐,正如六角环能够准确地叠在它们上方,此图是(a)图的拉普拉斯和低通过滤的图像;STM成像的参数是-263 mV,3.2 pA,77 K(摘自文献[10])Fig.2.Atomic structure of ice-clusters on Ru(0001).(a)Molecularly resolved crystalline cluster containing two water layers surrounded by exposed Ru.The blue dots in the inset show the preferred adsorption sites of third layer molecules within a hexagon.(b)The first and second layer are in registry with the underlying substrate,with the hexagonal rings precisely stacked above each other,as shown by the Laplace and low pass filtered image in(b).STM parameters:-263 mV,3.2 pA,77 K(Images are from Ref.[10]).
Nie等[23]发现可以在高偏压和小电流(-6 V,0.4 pA)的条件下对于多层冰进行无损伤的扫描隧道显微镜成像,进而观察到115—135 K温度下,4—5 nm厚度的冰层上最表面的冰在第二层冰岛表面的扩散和长大.日常生活中最常接触到的是六角相的冰,例如雪花,但低温下还存在着另一个结构的冰——立方相的冰.Thumer和Nie[25]在Pt(111)表面的多层冰的结构中发现螺旋位错的存在,这些螺旋位错的特性决定了Pt表面冰的结构是立方相还是六角相.但遗憾的是,这些研究都无法得到分子层次的氢键结构.
虽然STM实验需要导电的衬底,但是仍有两种办法可以利用STM研究绝缘体表面:一是在金属衬底表面生长超薄的绝缘体薄膜,这样来自针尖的电子仍然可以一定的概率透过绝缘体薄膜,隧穿到金属衬底;二是在绝缘体中进行掺杂,诱导出自由载流子.近年来,利用STM研究绝缘衬底上的水分子引起了很大的关注,主要集中在金属氧化物和碱金属卤化物[26-31].

图3 (a)Sr2RuO4表面在160 K下形成的单层水的STM图像;(b)重叠了衬底原子的放大STM成像;蓝色和红色的球分别表示Sr和O;水层由分解的OH基团(绿色的虚线圆)和水分子(黑色的虚线圆);OH吸附在Sr—Sr的桥位,但是水分子吸附在Sr的顶上;参数V=500 mV,I=100 pA(a);V=400 mV,I=100 pA(b)(摘自文献[24])Fig.3.(a)STM image of monolayer water grown on Sr2RuO4substrate at 160 K;(b)Zoom-in STM image with substrate atoms superimposed.Blue and red spheres denote Sr and O,respectively.The water overlayer is a mixture of dissociated OH groups(dashed green circle)and molecular H2O(dashed black circle). The OH adsorbed at the Sr—Sr bridge site,while the H2O was positioned on top of Sr.Set point:V=500 mV and I=100 pA(a);V=400 mV and I=100 pA(b)(Images are from Ref.[24]).
水吸附在金属氧化物表面比在金属表面更为复杂,因为水分子除了与金属原子成键以外还会和氧原子成键,这往往会导致水分子的分解.He等[32]在催化活性较高的anatase TiO2(101)表面吸附的单个水分子呈现“亮-暗-亮”的特征,结合密度泛函理论(density functional theory,DFT)发现水分子中的氧原子会和表面的Ti5c形成配位键,氢原子会和近邻桥位的氧原子形成两个氢键.另一方面,在rutile TiO2(110)表面上,水一般会吸附在Ti链上,并且会在氧空位上发生分解[33-35].在与TiO2(110)相似的RuO2(110)表面,水分子吸附在钌原子上,在238 K下沿着钌链扩散形成二聚体[36,37].当进一步升高温度到277 K,二聚体会进一步分解成H3O2和OH.在其他一些氧化物体系中人们也发现了水的分解,如图3所示,单层水吸附在Sr2RuO4表面会形成一个网络,它由锶原子上的水分子和在桥位的OH组成[24].水分子在FeO/Pt(111)表面130 K退火会形成大范围的二维岛[38],但是在羧基化的FeO表面,水分子在110 K倾向于和羧基形成氢键从而得到六角的纳米团簇.
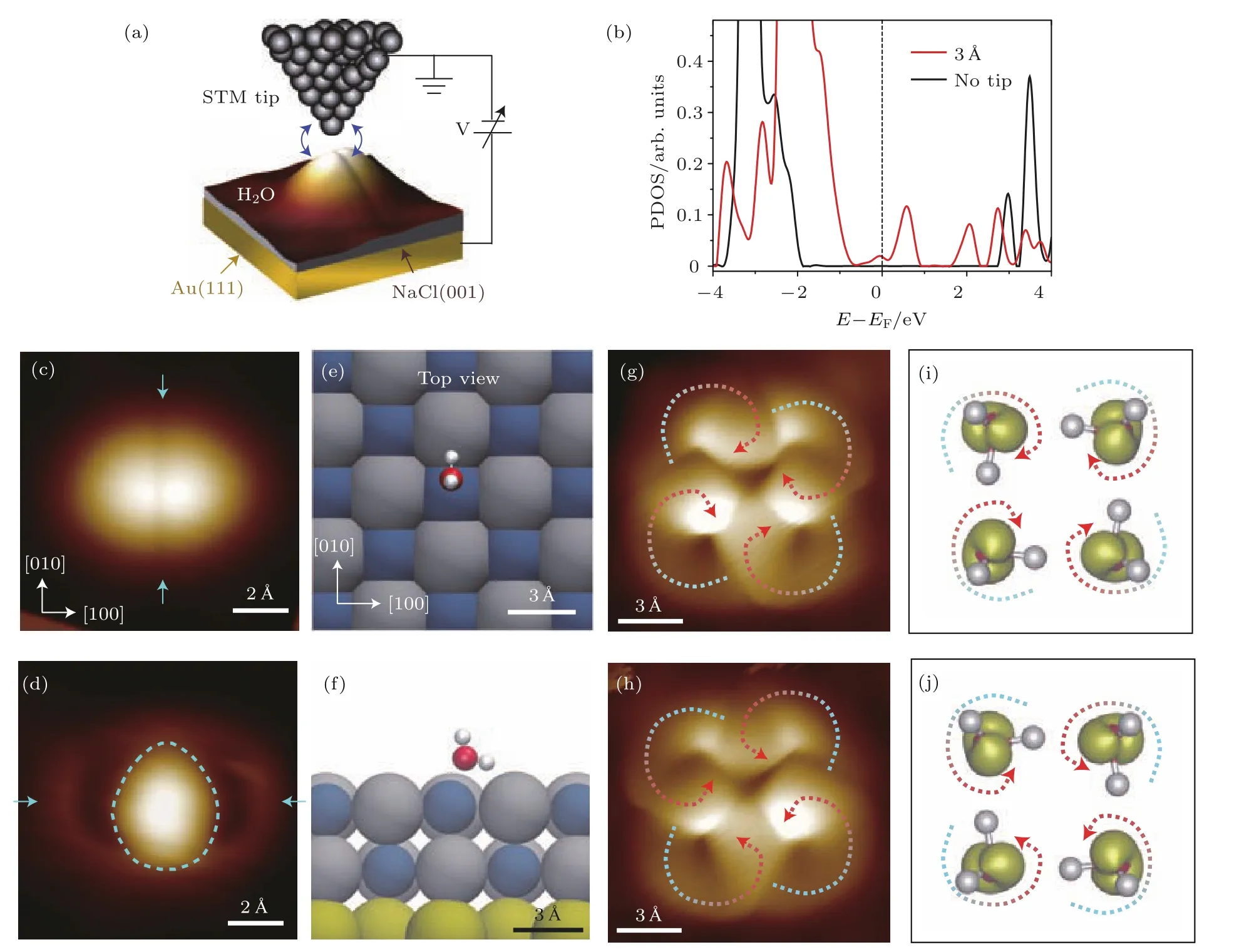
图4 NaCl(001)表面上单个水分子以及水分子四聚体的STM图像 (a)实验装置示意图,其中蓝色双箭头表示针尖与分子之间的耦合作用;(b)考虑针尖和不考虑针尖时计算得到的NaCl(001)表面单个水分子的投影态密度;(c),(d)分别为单个水分子的HOMO和LUMO轨道STM图像;(e),(f)单个水分子在NaCl(001)表面的吸附俯视图和侧视图;(g)和(h)两种具有不同氢键手性的水分子四聚体的HOMO轨道STM图像;(i),(j)由DFT得到的与(g)和(h)相对应的轨道图像(图摘自文献[28])Fig.4.STM images of water monomer and tetramer on NaCl(001):(a)Schematic of the experimental setup,the blue double arrows represent the coupling between the tip and water molecule;(b)projected density of states of water on NaCl(001)with and without the tip;(c)and(d)are the STM images of HOMO and LUMO of water,respectively;(e)and(f)are the top and side view of water adsorbed on NaCl(001)surface,respectively;(g)and(h)are the STM images(HOMO)of water tetramers with different chiralities,respectively;(i)and(j)are the orbital images by DFT which correspond to(g)and(h),respectively(Iamges are from Ref.[28]).
利用STM研究水分子的一个很大的限制是不能分辨水分子中的OH的指向,进而不能给出氢键网络的拓扑结构.近期,Guo等[28]成功地实现了在NaCl(001)/Au(111)表面上单个水以及水的四聚体的亚分子级分辨成像.该技术的关键是通过针尖-样品的相互作用来调控水分子的前线轨道,使得最高占据分子轨道(highest occupied molecular orbital,HOMO)和最低未占据分子轨道(lowest unoccupied molecular orbital,LUMO)往费米能级附近移动.这使得研究人员可以在零偏压附近对于水分子进行高分辨轨道成像,避免了高能隧穿电子对于分子的扰动.基于以上的轨道成像方法,单个水分子的空间取向可以确定下来,进而提供了在团簇结构中分辨氢键方向的可能.如图4所示,实验上发现不同的氢键方向可以导致两种手性的水四聚体.STM的超高空间分辨捕捉到了这两种结构转换,进一步分析发现质子协同隧穿在氢键结构转变中扮演着重要角色[39].此外,这种四聚体是NaCl(001)表面上二维冰的组成基元[40],形成不同于传统六角bilayer冰的结构,其中包含了高浓度的Bjerrum D-type缺陷.
2.2 非弹性电子隧道谱技术
除了氢键的空间几何构型,水分子之间的氢键强度、氢键动力学、同位素效应等问题也是水科学领域研究的重点.基于STM的非弹性电子隧道谱(inelastic electron tunneling spectroscopy,IETS)是研究这些问题的一个有效手段.该技术的核心思想是通过高度局域化电子的非弹性隧穿来激发单个水分子的振动,从而获取单分子尺度上的振动模式信息.

图5 (a)—(c)I-V,dI/dV和d2I/dV2的示例图,非弹性电子隧穿的信号出现在阈值电压hω/e,ω是振动的角频率;(d)—(f)是非弹性过程的示意图(图摘自文献[41])Fig.5. (a)–(c)schematic spectra of I-V,dI/dV,and d2I/dV2,respectively,showing the inelastic electron tunneling features at the threshold bias voltage hω/e, ω is angular frequency of the vibration;(d)–(f)schematic diagram of the inelastic electron tunneling process(Images are from Ref.[41]).
2.2.1 IETS技术简介
从隧穿电流中得到振动信号这个想法最早是由Jaklevic和Lambe[42]在金属-氧化物-金属隧穿结中实现的.Stipe等[43,44]在30年以后将该技术应用到STM中,首次获得了单分子级别的分子振动谱.IETS的基本原理如图5,当隧穿电子的能量足以激发分子的振动时,会打开新的非弹性隧穿通道,这直接导致了电流的增大.但是非弹性电流相较于弹性隧穿电流还是极其微弱的,为了更精确地分离出非弹性电流的贡献,通常要对总电流做偏压的二阶微分,测量电流的二阶微分信号随偏压的变化,就得到了IETS.由于非弹性激发只依赖于电子能量的大小而与电流的方向无关,所以在正负偏压都会得到IETS信号,从而形成关于原点中心对称的谱图,这个特征往往被当作判断是否为非弹性激发的标准.
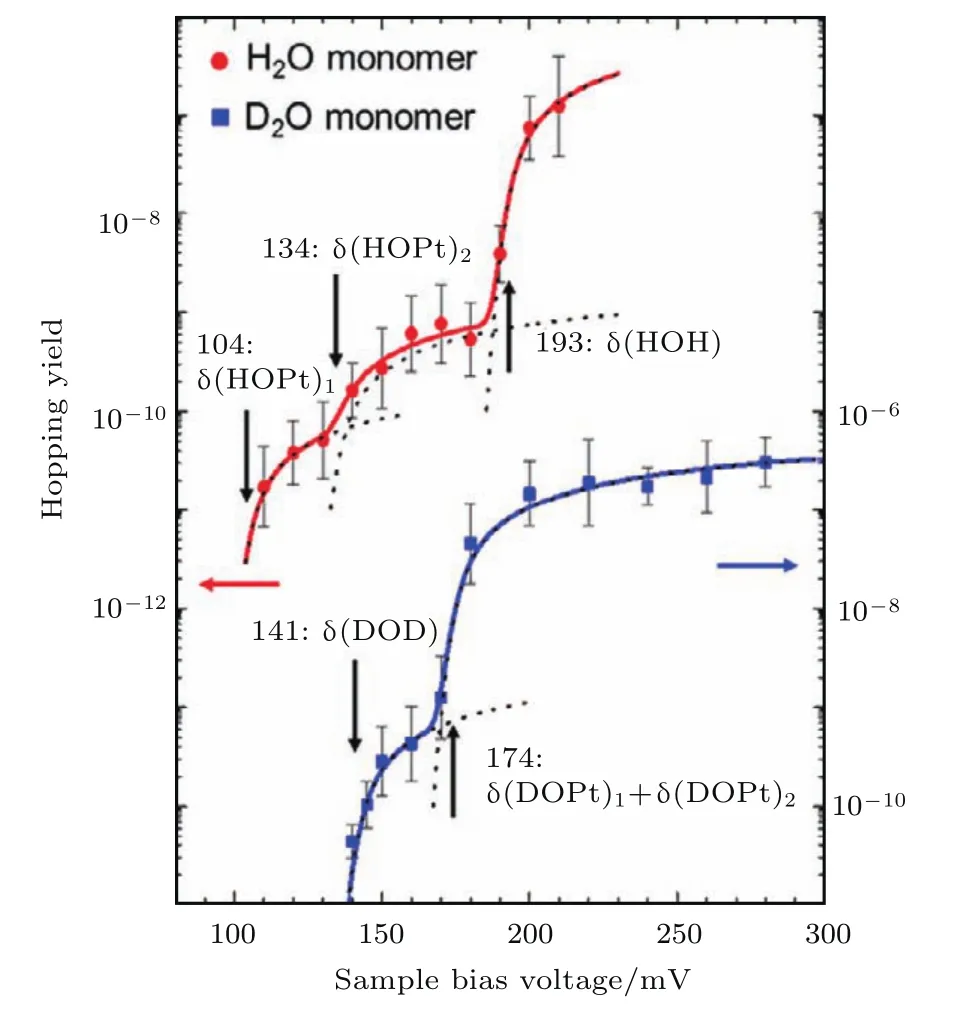
图6 H2O和D2O单个水分子在Pt(111)表面侧向跳跃的STM-AS谱线以及对应的谱图拟合 红圈和蓝色方块分别代表H2O和D2O的实验结果,粗实线代表最佳的拟合,虚线代表分段拟合的部分Y(V)tot(摘自文献[45])Fig.6.STM-AS and spectral fit of the lateral hopping of H2O and D2O monomers on Pt(111).The red circles and blue squares represent the experimental results of STM-AS for H2O and D2O,respectively.The thick solid curves represent the best- fit spectra,and the broken curves represent the fraction of simulated Y(V)tot(Images are from Ref.[45]).
2.2.2 STM-IETS技术在水-固界面的应用
Morgenstern和Nieminen[46]最早获得了在77 K下Ag(111)表面水的IETS.因为分子轨道都远离费米能级,所以他们得到的是非共振IETS,信噪比非常低,无法准确给出每个共振峰对应的振动模式.在另外一个实验中,Kumagai等[47]可以测量Cu(110)表面上单个羧基分子以及团簇的IETS.OH/OD的弯曲模式和伸缩模式都可以清晰地从谱图中确定,这些模式被进一步用来研究羧基分子的动力学.但是非弹性激发导致羧基的结构发生变化,进而使得IETS谱图过于复杂.
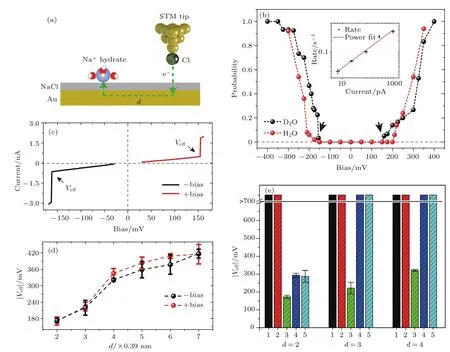
图7 针尖诱导的Na+水合物的动力学过程 (a)STM针尖诱导的在针尖距离为d×NaCl(001)晶格常数处的Na+水合物非弹性电子激发的示意图,金衬底在其中作为媒介,NaCl(001)晶格常数为0.39 nm;(b)在距离CO针尖d=4的条件下,Na+·3D2O和Na+·3H2O的扩散概率与电压依赖关系,电压的持续时间是1.2 s,且扩散的概率是从50个事件中统计得出,插图展示了在d=2的条件下CO针尖170 mV电压下Na+·3D2O的扩散概率随电流的依赖关系,实线是对于数据用指数关系的最小二乘法拟合,R∝IN,其中N=1.02±0.08,可见是一个单电子激发;(c)Na+·3D2O在距离Cl-针尖d=2的条件下的电压电流关系,电流会在Veff处有一个跳变;(d)在Cl-针尖的条件下,Na+·3D2O的正负偏压的Veff对于距离依赖关系的一致性;(e)在d=2,3,4条件下,对于不同的Na+·nD2O(n=1—5)的Veff的比较(摘自文献[48])Fig.7.Tip-induced diffusion dynamics of Na+hydrates.(a)Schematic diagram of the Au-mediated inelastic electron excitation of the Na+hydrates with the STM tip at a lateral distance of d×the lattice constant of NaCl(001),which is 0.39 nm.(b)Bias dependence of the diffusion probability of Na+·3D2O and Na+·3H2O with a CO tip at d=4.The voltage pulse duration for each event is 1.2 s.The diffusion probability is a statistics from 50 events.The inset shows the current dependence of the diffusion rate of Na+·3 D2O with a CO tip at d=2 under 170 mV.The solid line is the least-squares fit to the data with a power law,R∝IN,where N=1.02±0.08,indicating a one-electron process.(c)Current-bias relationship of Na+·3D2O with a Cl- tip at d=2,where the current jumps occur at Veff.(d)Lateral distance dependence of the positive(red)and negative(black)Vefffor Na+·3D2O with a Cl- tip.(e)Comparison of Vefffor Na+ ·nD2O(n=1–5)at d=2,3 and 4(Images are from Ref.[48]).
虽然非弹性激发导致的结构失稳使IETS谱图变得复杂,但是也给人们利用STM实现action spectroscopy(AS)提供了机会.2004年,Kawai等[49]率先将光谱学中的AS引入到STM中.在反应产额(例如:扩散、转动、断键等)随偏压的关系图中,与反应相关的振动激发和电子激发都会呈现对应的电压阈值[2,50-52],如图6所示.通常认为IETS谱会受限于Propensity rule[53-55],也即特定的振动模式是否能被激发需要考虑到前沿轨道的对称性,但是AS可以突破这个限制,探测许多IETS中禁阻的振动模式.此外,IETS会涉及到弹性隧穿通道与非弹性隧穿通道之间的竞争,而AS谱图则不受弹性隧穿的影响,其解释相较于IETS会容易许多.
作为一种与IETS-STM互补的手段,STM-AS已经被成功运用到许多水相关的体系中,例如:单个水分子的选择性分解[26]、氢键的辨别[45]、氢原子转移路径中的识别[56-58].近期,Peng等[48]利用AS发现了水合钠离子在NaCl(001)表面扩散的幻数效应.如图7所示,通过STM的非弹性激发可以诱导水合钠离子的横向扩散,包含三个水分子的钠离子水合物对应的阈值偏压总是比其他钠离子水合物小.通过详细的理论计算,他们发现离子水合物与衬底表面的对称性匹配程度导致了三个水分子的水合物扩散势垒更低,扩散速度更快,对应于一种全新的动力学幻数效应.
另一方面,通常的IETS信号只有百分之几的微分电导变化[41],这对于STM系统的机械稳定性要求极高.为了进一步提高信噪比,提升水分子的非弹性散射截面,Guo等[59]发展了针尖增强的STM-IETS技术.他们首先利用绝缘的NaCl薄层减少了水分子和金衬底的耦合,使得电子可以在分子中停留更长的时间,提高了电-声作用的概率.其次,他们利用氯原子吸附的STM针尖调节分子前线轨道与费米能级之间的能量差,从而有效增加费米能级附近的电子态密度,从而实现了水分子的近共振激发IETS.从图8中可以看到针尖增强的IETS信噪比非常高,相对电导变化接近30%,比传统IETS提高了一个量级.因此,利用针尖增强IETS可以非常精确地识别水分子的不同振动模式(包括拉伸、弯曲、转动等).得益于它的超高精度,实验人员甚至可以从OH/OD伸缩模式的红移定量得到氢键的强度[60].值得一提的是,他们进一步通过H/D的同位素替代实验给出了单键尺度上核量子效应对氢键强度的影响,澄清了氢键的量子本质[61].

图8 单个水分子针尖增强的非弹性隧道谱 (a)实验体系示意图,单个水分子(D2O)竖直吸附在NaCl(001)/Au(111)衬底上;红色、白色、金色、绿色、紫色小球分别代表O,D,Au,Cl-和Na+;(b)针尖增强的IETS信号的原理示意图;(c)单个水分子的dI/dV及对应的d2I/dV2谱线;灰色曲线表示Cl针尖高度为-1.2 Å时NaCl表面的信号;蓝色曲线是Cl针尖高度为-0.4 Å时水分子上的信号;红色曲线是Cl针尖高度为-1.2 Å时水分子上的信号;谱线上的出现水分子的特征振动信号:“R”(rotational),“B”(bending),“S” (stretching)(摘自文献[59])Fig.8.Tip enhanced IETS of single water molecule.(a)Schematic of the experiment setup.Single water(D2O)adsorbes vertically on the NaCl(001)/Au(111).O,D,Au,Cl-,and Na+are denoted by red,white,golden,green,and purple spheres,respectively.(b)Schematic of the tip enhanced IETS.(c)dI/dV and d2I/dV2 spectra taken at different tip heights.Red(–1.2 Å)and blue(–0.4 Å)curves were taken on the D2O monomer.Gray curves(-1.2 Å)were acquired on the NaCl surface(denoted as “bkgd”).The vibrational IET features are denoted as“R”(rotational),“B”(bending),and “S”(stretching)(Images are from Ref.[59]).
2.3 电化学扫描隧道显微镜
水和金属的相互作用在诸如多相催化、电化学中发挥着重要作用,如电极表面水分解产生氢气和氧气的过程.因此对水在金属表面的行为研究尤为重要.STM自发明以来,由于其具有较高的原子分辨率,迅速成为了研究人员必备的纳米表征工具.STM起初只是在真空条件下的固-气界面上应用,但水的浸润过程和金属的腐蚀等许多有趣的实验现象和工业相关的现象发生在固-液界面上.电化学扫描隧道显微镜(electrochemical scanning tunneling microscopy,ECSTM)的发明,能够原位得到固-液两相的反应信息,同时具有原子分辨的能力.
2.3.1 EC-STM简介
EC-STM能够在原子尺度上对电极表面进行原位实空间成像,同时得到固-液界面的信息,因此迅速成为解析电极表面结构和研究固-液界面上动力学过程的重要工具.1986年,Sonnenfeldh和Hansma[62]最早利用STM获得了水和水基电解液中石墨和金膜的结构信息.然而早期实验只有两个电极(针尖和样品电极),无法控制电极电位.由于针尖在装置中也起到电极的作用,所以在尖端会有极化效应和电化学的电荷转移反应,因此需要良好稳定的针尖环境.1988年,Siegenthaler等[63]引入了参比电极(例如Ag/AgCl)组成了三电极体系,通过双恒电位同时控制两个电极电位.后来许多研究组(Wandlowski[64],Weaver[65])也做出了改进性的工作.
图9是EC-STM的工作原理图,它由STM部分和电化学部分组成.电化学部分由三电极系统组成,分别是工作电极、参比电极、对电极,三者置于电解池中.通过双恒电位仪控制针尖和样品的电极电位,从而发生电化学过程,整个过程由STM记录下来.STM部分中针尖也是另一个工作电极,同时组成了第二个三电极系统.通过前置放大器放大电流,得到样品表面形貌信息.
通常在EC-STM中,流过针尖的电流有几个分量:1)STM隧穿电流;2)针尖/电解液之间电化学反应的法拉第电流;3)针尖/电解液界面双电层之间发生的充放电电流.法拉第电流和充放电电流要比隧道电流的设定值高(隧道电流是nA数量级,典型的法拉第电流是mA数量级),如果不采取适当的措施,二者将会以不同的方式影响STM的成像结果(有时甚至不能成像)以及STM图像的重复性,而且还会使STM的实验数据难以解释.
因此,EC-STM工作中要考虑的是设法抑制和隔离针尖上的法拉第电流和充放电电流,目前解决这一问题的方法有两种:一是利用探针封装技术[66],可以做到将针尖金属侧面覆盖有机高聚物等绝缘体而只露出尽可能小的探针金属尖端,这样可以产生隧道电流的同时又可以极大地抑制法拉第电流和充放电电流;二是利用Itaya等[67]发展起来的四电极系统,它相当大的程度上克服了STM在电化学研究上的困难.

图9 EC-STM工作原理示意图,双恒电位控制针尖和样品的电位Fig.9.EC-STM con figuration,the bipotentiostat controls potential of tip and sample with respect to reference electrode.
电解液的隧穿机理现在还不是很清楚,仍需要进一步进行研究.与在真空中的隧穿过程不同,在电解液中的隧穿势垒要比真空中低.一方面的解释是金属表面由于吸附了水而功函降低;另一方面原因是由于水分子的氢原子和氧原子的运动和分布,金属表面势能出现二维网络极值的振荡,而不再是常数[68].
EC-STM自发明以来,在诸多领域发挥着不可替代的作用,如:研究电极表面结构、吸附质层的结构,研究吸附动力学、生长机理和表面相转变等.例如EC-STM在理解腐蚀、沉积、吸附等过程有着重要的作用.腐蚀过程包括了表面溶解、形成钝化表面和缓蚀等.利用EC-STM已经研究了许多材料在多种条件下的腐蚀,包括Cu,Ni,Fe等金属.通常钝化层的缺陷位或者金属边处氧化层比较薄,这些可能是腐蚀的起始位置.2012年,Ye等[69]研究了不锈钢局域腐蚀过程.他们首次观察了腐蚀活性位点和惰性位点的垂直形貌,并且讨论了产生这种差异的原因有可能和电场的分布、Fe3+/Fe2+的浓度有关.
2.3.2 EC-STM在水-金属界面探测的应用
固-液界面的研究在基础科学和工业应用等方面有着广泛的应用.固-液界面分别由固体、吸附水层和双电层等部分组成.构成固-液界面的材料可以是金属、合金、半导体等;溶液中可以包含水分子、水合离子、有机分子等.其中,水-金属界面作为固-液界面的一种,尤其是和产氢、储能和转化等燃料电池相关,在电化学方面引起了广泛的关注.当充电电极接触到水溶液时,极性分子或者离子会被静电力吸引到电极表面,于是两相处会产生电势差,形成双电层.双电层由电子、溶剂分子、溶剂化离子等组成.
人们对双电层的认识是一步步加深的.1853年,Helmholtz[70]提出双电层模型,将双电层和平行板电容器类比,认为吸附极性分子或者离子紧密排布在极板上.该模型较适合于浓电解质的情况,但是不适合随极板距离有电荷分布的情况.随后经过许多人的不断改进,1963年,Bockris,Devanthan和Müller提出了新的双电层模型,即目前最具代表性的BMD模型[71].
图10是BMD模型的示意图.如图所示,在电解液中,第一层水分子紧密定向排列在金属的表面,形成内亥姆霍兹平面(inner Helmholtz plane,IHP);第二层水分子部分定向排列,水分子和离子会形成溶剂化离子,共同形成外亥姆霍兹平面(outer Helmholtz plane,OHP).继续向外是扩散层区间,这里面有溶剂化离子,此时和电极之间的相互作用仅仅是远程的静电力,最外面是溶液本体.
金属电极表面水的吸附构型对理解催化过程有着重要的意义.H2O和H3O+有助于稳定阴离子,减小它们之间的静电排斥力;同时有助于连接阴离子形成二维结构.2000年,德州农工大学的Kim等[72]利用EC-STM研究了硫酸溶液里面的Pd(111)表面阴离子吸附的稳定构型.他们利用原子级尖锐的STM针尖得到了高质量的图像,并且从原子尺度上分析表面高度致密的分子内和分子间结构.如图11(a)所示,H3O+和H2O和SO2-4被明确的区分出来,并且归属了H2O层和H3O+层.

图10 双电层模型(BMD model) 1,内亥姆霍兹平面;2,外亥姆霍兹平面;3,扩散层;4,溶剂化离子;5,特殊吸附离子;6,电解液溶剂分子Fig.10.Schematic representation of a double layer on an electrode(BMD)model.1,Inner Helmholtz plane(IHP);2,outer Helmholtz plane(OHP);3,diffuse layer;4,solvated ions(cations);5,speci fically adsorbed ions(redox ion,which contributes to the pseudocapacitance);6,molecules of the electrolyte solvent.
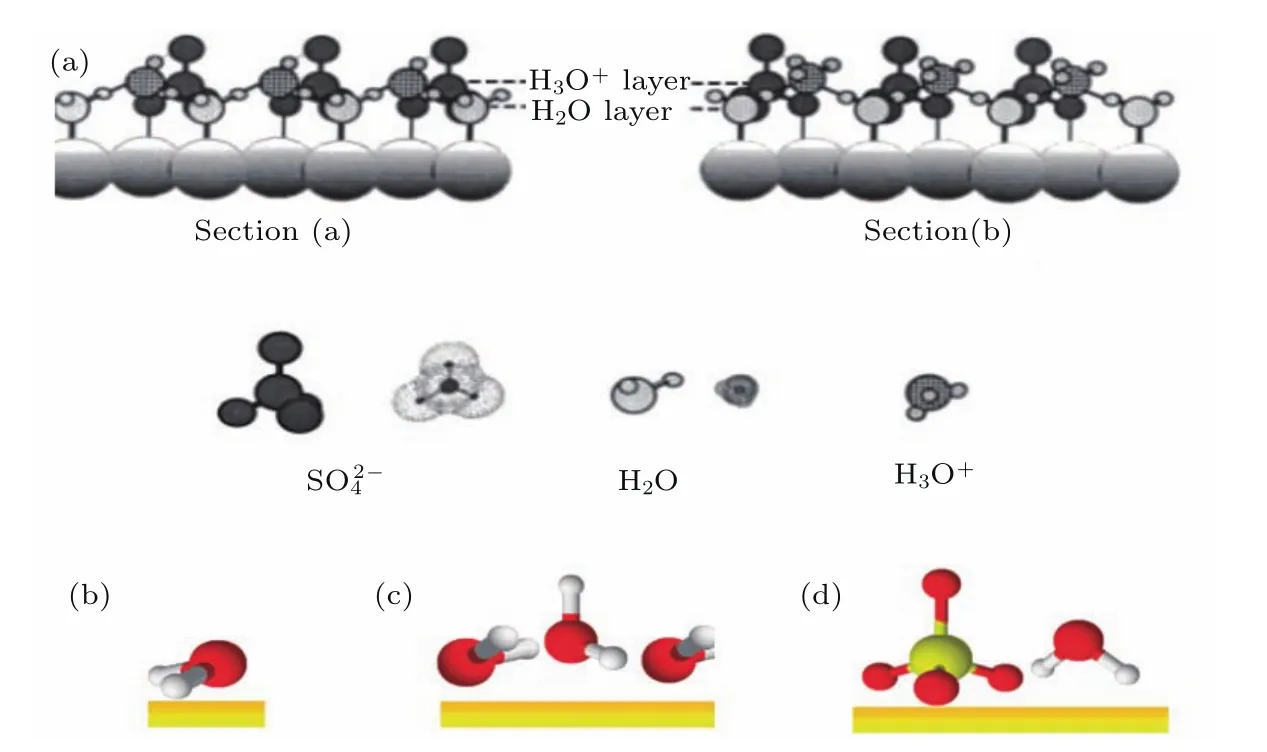
图11 硫酸溶液中水分子离子吸附结构图 (a)Pd(111)上H3O+,H2O和吸附结构侧视图;(b)—(d)金电极上水的吸附结构模型(摘自文献[72,73])Fig.11.(a)Side view of structural model of the Pd(111)/H3O+/H2O on Pd(111);(b)–(d)proposed model structures of water adsorbed on gold electrodes(Images are from Refs.[72,73]).
2011年,莱顿大学的Garcia-Araez等[73]研究了金电极表面受电势控制的硫酸水溶液中水吸附结构.如图11(b)—(d),他们利用表面增强红外吸收谱(surface-enhanced infrared absorption spectroscopy,SEIRAS)发现水吸附受到电极电势的影响.在0
理论计算对水-金属界面进行了大量的研究.2006年,Schnur和Gross[74]认为在六方对称的过渡金属上水的吸附为双层结构,这种结构类似于致密的冰.在这种结构中,如图12(a)—(c),第一层水分子和金属面平行,它的氧在金属的top位上;对于其他的分子,有两种不同的取向,分别是H-up和H-down结构,它们的氢原子分别远离或者朝向金属面.但是对于像钌、铑、镍等和水分子作用强烈的金属,靠近金属的水分子会解离成H和OH,这种结构是更加稳定的.有研究认为金属电极吸附水后功函会减小[75].作者同时发现吸附水构型的改变会对金属电极的功函数产生重要的影响.例如,H-up和H-down两种构型由于相反的偶极符号对功函的响应不同.从图12(d)中可以看出,H-up和H-down两种构型对不同金属的功函差大概均为2 eV.对于钌,水的构型使用的是前述的水解离的结构.

图12 (a)H-down双层结构;(b)H-up双层结构;(c)半解离的水-羟基双层结构;(d)H-down,H-up和半解离结构在金属表面引起的功函的变化(图摘自文献[74])Fig.12.(a)H-down bilayer;(b)H-up water bilayer;(c)half dissociated water-OH-bilayer with the additional hydrogen atoms at the center of the hexagonal rings;(d)calculated work function change induced by the presence of H-up H-down and half-dissociated water bilayers on metals(Images are from Ref.[74]).
2007年,Sugino等[76]利用effective screening medium(ESM)研究了施加了负偏压的水-铂金属界面.研究表明,带电金属表面的水分子会形成严格的层状结构,其中氢原子会指向表面,从而增加屏蔽能力.
EC-STM为STM提供了电化学的环境,为在水溶液中研究电子隧穿过程创造了条件.和真空中不同,隧道结内填充着水而不是真空,电子隧穿的势垒和隧穿距离会发生明显的变化.1995年,亚利桑那州立大学的Vaught等[77]研究了在Pt-Ir针尖和Au(111)表面水层间距离和隧穿电阻的关系.如图13(a)所示,隧穿电阻和距离呈现非指数依赖关系,这和在超高真空环境中观察到的现象呈现鲜明的对比.他们猜测其中一个原因是在针尖靠近样品时水分子的构型发生了变化.1998年,浦项科技大学的Hong等[78]研究表明,隧穿势垒高度强烈的依赖于极性.图13(b)表明在正的样品偏压和高电场的情况下,样品表面第一层水分子被极化,水分子中的氧朝下,并且侧向方向压缩,因此隧穿概率会减小.随着偏压的增加,氧原子密度会增加,同时隧穿概率会进一步下降;样品加负偏压,表面的水层会反向极化,这样隧穿会更加容易.但是这种隧穿的不对称并不能完全用水的取向改变去解释,因为如果STM涉及到两个电极,如果两个电极上水分子的行为相同,那么隧穿始终是对称的.隧穿的不对称有可能还和STM针尖的形状有关,在针尖尖端,水分子可能排列成了不同的形状.

图13 (a)隧道电阻的对数(Log10)和针尖样品距离的关系;(b)正负样品偏压时表面水层的排列,正偏压侧向方向水层压缩,负样品偏压水层结构开放(摘自文献[77])Fig.13.(a)Log(base 10)of the tunneling resistance plotted against the distance the tip moved from the position at which R=109V;(b)pictorial illustration of the water monolayer arrangement on a positively and negatively biased sample surface which will lateral compression or be more open structure(Images are from Ref.[77]).
EC-STM在催化领域发挥着重要的作用,其中析氢反应和氧还原反应由于在产氢、储能等方面的巨大的应用而受到广泛的关注.2017年,P fisterer等[79]利用EC-STM研究了异质催化剂中活性位点的问题.作者使用了单原子层Pd岛覆盖的Au(111),如图14(a)和图14(b),当STM针尖扫过台面时,此时没有析氢反应发生,隧道电流噪音水平近似一样;当针尖越过台阶时,析氢反应发生,此时隧道电流上出现了明显不同的噪音.图14(c)是大气条件下硫酸浓度为0.1 M时Au/Pd的边界,调节电压在Pd上发生了析氢反应,在活性较低的Au上不发生析氢反应.从图14(d)中可以看出,在边界处催化活性明显变化,因此利用水的析氢反应证明了异相催化剂活性位点的问题.
EC-STM兼具电化学环境和STM原子分辨的优势,在水科学研究中水吸附构型、水极化、水催化等领域具有重要应用,也能进一步帮助解决水科学的基础研究和工业应用等方面问题.
2.4 非接触式原子力显微镜
STM使得表面物理化学领域的研究达到了一个新的高度,并且带动了例如EC-STM等检测工具的发展.STM的原理是利用隧道电流,其缺点是只适用于导电样品.而且要想在原子尺度上研究水,STM针尖逼近样品时,会对水的微观结构造成微扰.因此,需要一种非侵扰式的工具来对水进一步研究.1986年,Binnig,Quate和Gerber三位科学家共同发明了原子力显微镜(atomic force microscope,AFM)[80].它的原理是利用柔性悬臂梁感知针尖和样品之间的相互作用力.进一步发明的动态非接触AFM模式(non-contact AFM,NC-AFM),使得AFM的分辨率进一步提升.
2.4.1 NC-AFM简介
AFM通过末端黏有尖锐针尖的悬臂梁扫过样品表面,通过悬臂梁的弯曲来间接获得形貌信息.AFM有两种不同的工作模式,接触模式(静态模式)和非接触式模式(动态模式),即NC-AFM[81].NC-AFM悬臂是由机械激励驱动其在共振频率(f0)下振动,它的振幅(A)一般在10 nm以下,针尖可以在距离样品相对较近的情况下稳定工作,且针尖不会撞到样品表面,故不会对样品表面造成损害.
NC-AFM分为振幅调制(AM)和频率调制(FM)两种[82].AM-AFM中是以固定的频率和固定的振幅产生激励,当针尖靠近样品时,由于针尖和样品相互作用,悬臂梁的振幅会发生变化.因此,振幅可以用来作为样品表面成像的反馈信号.在AM模式中,振幅响应时间τ≈2Q/f0,其中品质因子Q值代表振动的稳定性,Q值越大,每一次振动中能量损耗越小.在真空中Q值达到105后,AM模式信噪比有所提高,但是扫描速度会非常慢.所以AM模式在真空中不大适用.而FM-AFM则解决了这个问题,它的反馈维持振幅恒定,测量针尖和样品之间作用力梯度引起的共振频率的偏移(∆f).AM模式主要在大气和液相中工作,而FM模式应用于超高真空的环境中,两种模式均为NC-AFM的发展做出了重要的贡献.

图14 (a),(b)检测活性位点的图示,当针尖和样品的环境改变时,如样品表面(a)台面(b)台阶时,隧穿势垒随时间发生变化,在这种情况下,当针尖越过台阶时相对在台面上扫描时电流噪音更可能出现;(c)0.1 M硫酸条件下发生析氢反应时在Au(111)Pd岛的边界的STM图,插图是Au(111)基底的原子分辨;(d)是(c)图中每条线扫描的细节图(摘自文献[79])Fig.14.(a),(b)A scheme explaining the concept.When the local environment between the STM tip and the sample changes(in this case,when the tip is over a terrace,(a)versus a step,(b)in the sample),the tunneling barrier also changes over time,in a way that is driven by the changes in approaching and departing reactants and products.In this scenario,increased tunneling-current noise is likely to appear when the tip is over a step edge,which is more active than the terrace sites.(c)An STM image of the boundary between a Pd island and the Au(111)substrate under HER conditions in 0.1 M sulfuric acid(constant-height mode).The inset shows an atomically resolved image of the Au(111)substrate.(d)Detailed STM line scans for the case shown in(c)(Images are from Ref.[79]).

图15 针尖和样品相互作用力随距离变化示意图Fig.15.Total interaction force again distance between tip and sample.
NC-AFM利用针尖和样品相互作用力来进行成像.如图15,针尖和样品之间由吸引力和排斥力成分组成.吸引力主要有范德瓦耳斯力、静电力和化学吸引力,排斥力主要是Pauli排斥力.范德瓦耳斯力来源于原子和原子之间的局域瞬时偶极作用;针尖和样品之间的电势差或功函差可以产生静电力,这两者认为是长程力,短程力有短程化学吸引力和Pauli排斥力.一般认为,长程力没有原子成像的能力,只是成像的背景力,要得到高分辨的成像必须要针尖逼近到离被测样品的表面非常近的地方.
图16是NC-AFM的信号检测原理的框图[83],它由振幅控制模块和频率测量模块组成.悬臂发生偏转后产生信号进入带通滤波器,然后分别进入锁相环(PLL)、相位调节器和交流直流转换器.基于PLL的频率调制解调器测量频率偏移信号,并将其转化为电压信号.相位调节器调节悬臂振动激励信号和悬臂振动信号相位相差π/2,使激励信号最小.交流直流转化器将悬臂偏转振幅转化为直流信号,与振幅设定值比较后提取能量耗散信号.相位调节器和交流直流转换器共同组成振幅控制模块.NC-AFM成像模式由频率偏移的反馈开与关可以分为恒频率偏移模式和恒高度模式.恒频率偏移时,反馈打开通过实时记录压电陶瓷管的高度得到表面形貌信息;恒高度模式时,反馈关闭后针尖和样品距离不变,记录频率偏移信号得到恒定高度下频率偏移图.
1998年,德国雷根斯堡大学的Giessibl发明了qPlus传感器.它由具有压电自检测的石英音叉作为力传感器,其中一个悬臂固定,另一个悬臂处于自由状态,末端黏有针尖提取隧道电流信息,同时可以得到力的信号.石英音叉弹性常数k一般为1800 N·m-1,共振频率为32—200 kHz.相比于传统的激光检测的Si悬臂来说具有比较高的Q值,并且石英音叉硬度相对较大,针尖可以距离样品更近,可以得到样品分子内或者分子间的信息.
qPlus传感器在成像分辨率上具有极大的优势,推动了NC-AFM在原子尺度上化学成像的发展.2009年,苏黎世IBM实验室的Gross等[84]利用CO修饰的针尖在5 K、恒高度模式下得到了并五苯分子的高分辨图像,这是科学家第一次在实空间得到分子的化学结构信息.图17(a)是并五苯分子的“球棍模型”,图17(b)和图17(c)分别是吸附在Cu(111)上并五苯分子的STM和频率偏移图像.从AFM图可以看出,并五苯分子的每个六元环都清晰可见,甚至能够对C—H键进行成像.并五苯分子一圈周围的黑色区域作者认为是范德瓦耳斯力的背景,而亮色衬度是由于CO针尖和分子骨架产生的Pauli排斥力.

图16 NC-AFM的信号检测原理框图(恒定振幅控制和频率偏移控制)(摘自文献[83])Fig.16.Block diagram of the non-contact AFM feedback loop for constant amplitude control and frequency-shift measurement(Images are from Ref.[83]).
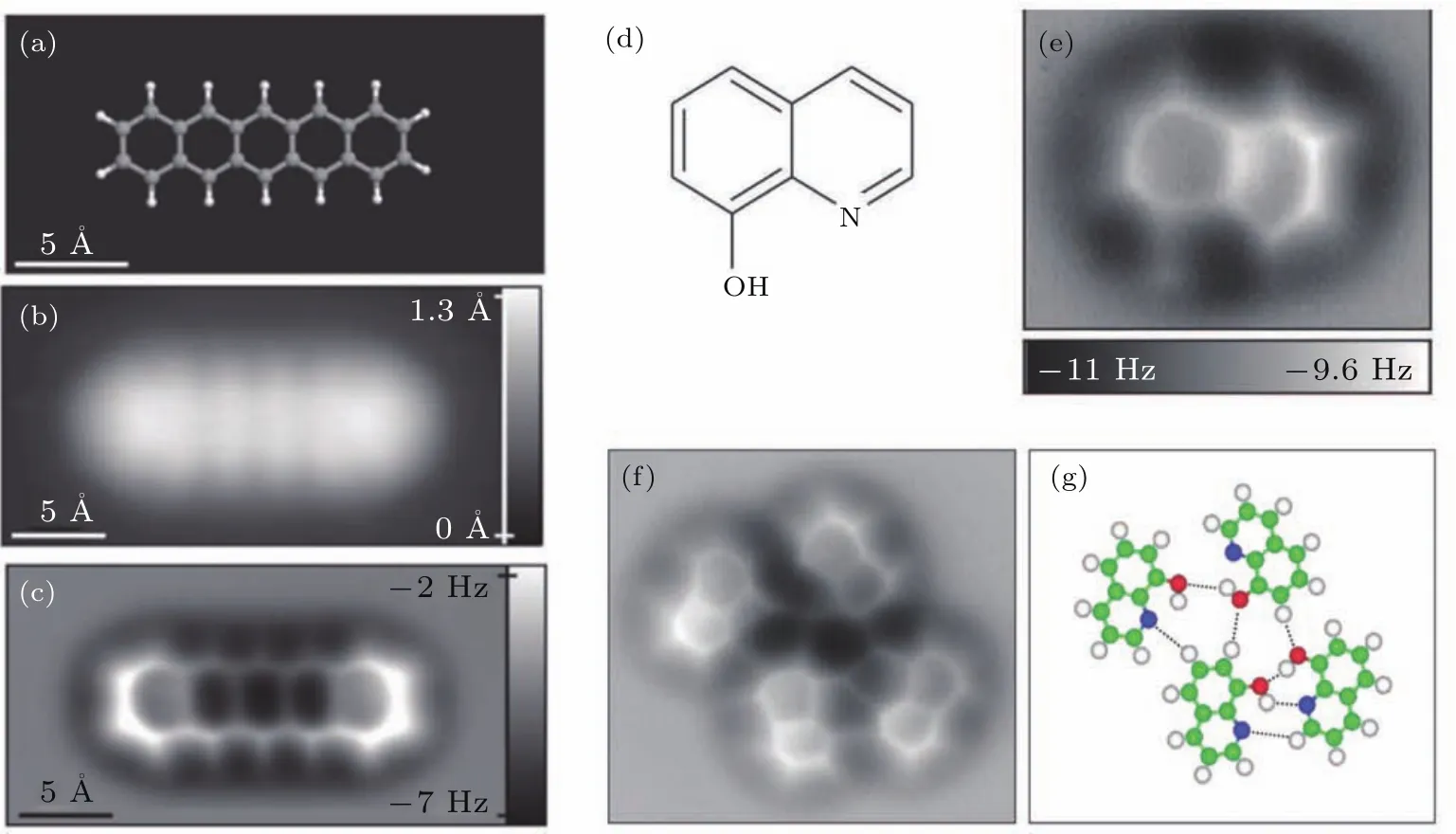
图17 (a)并五苯分子的球棍模型;(b)STM恒流图像;(c)CO修饰的针尖获得的并五苯分子恒高AFM图;(d)8-羟基喹啉的化学结构;(e)单个分子的恒高AFM图;(f)四聚体的恒高AFM图;(g)分子间类似氢键结构,碳、氮、氧、氢分别为绿色、蓝色、红色、白色.(图摘自文献[84])Fig.17.(a)Ball-and-stick model of the pentacene molecule;(b)constant-current STM;(c)constant-height AFM images of pentacene acquired with a CO-modi fied tip;(d)chemical structure of 8-hq;(e)constant-height AFM frequency shift images;(f)constant-height frequency shift images of typical molecule-assembled clusters and their corresponding structure models;(g)indicate likely H bonds between 8-hq molecules.Green,carbon;blue,nitrogen;red,oxygen;white,hydrogen(Images are from Ref.[84]).
除了可以对分子骨架进行成像,NC-AFM还可以对分子间相互作用等弱相互作用力进行识别.国家纳米科学中心的仉君等以8-羟基喹啉为研究体系,得到了在Cu(111)上氢键连接的四聚体的高分辨图案.如图17(d)—(g),8-羟基喹啉在CO针尖下清晰成像出分子骨架,同时形成的四聚体结构中,利用NC-AFM可以对其分子间相互作用力进行成像,他们认为氢键处电荷密度增大是成像的主要原因.目前关于氢键成像存在着争议[85,86].正是由于争议的存在,使得人们对NC-AFM成像的机理愈发重视.
2.4.2 NC-AFM在表/界面水领域的新进展
NC-AFM的迅猛发展在水科学领域也发挥了重要的作用.NC-AFM为界面水提供了高的水平分辨率,但是针尖靠近样品时,要控制针尖对水层的微扰.在室温大气环境下,针尖靠近样品时,针尖上的水和样品表面的相互作用,会导致测量误差.基于NC-AFM的静电力显微镜(scanning polarization force microscopy,SPFM)[87,88],利用静电的相互作用,施加了偏压的针尖距离样品表面10—20 nm,避免了针尖和样品之间的接触.
1995年,胡钧等[89]利用发明的SPFM研究了室温下云母片上水的凝聚和蒸发过程.利用SPFM可以对两个过程进行直接成像.凝聚过程中水相可以分为两种,25%湿度时,形成直径小于1000 Å的二维水团簇;高于25%的湿度时,形成大的二维岛状水层,这个岛状水层和云母的晶格相关;随着湿度的增加,岛状生长会在湿度为45%时完成.水的蒸发过程同样可以观测到.如图18,对于岛状的水层,作者发现针尖和样品接触后会诱导接触点处水的凝聚.针尖退回后,通过SPFM可以对过量的水形成的分子层厚的岛进行成像.水岛的边界经常是角度为120◦的多边形,通过比较云母片的晶格,可以得到边界的方向和云母的晶体学方向相关.因此作者得到了分子层厚的水层有着与冰类似结构的结论,即“室温下的冰”.
2011年,Santos和Verdauer[90]利用AM模式的AFM测量了水层的高度.在研究中,样品表面分为干燥和润湿区域.作者考虑了三者相互作用的区域,Wnc,Wc和排斥力区域.Wnc对应于针尖和样品的水层都没有被扰动的情况;Wc定义为水层被扰动,但是针尖和样品没有接触;排斥力为针尖和样品接触.通过实验和模拟在Wnc区域无论是干燥和润湿区域得到的是水层的真实高度.
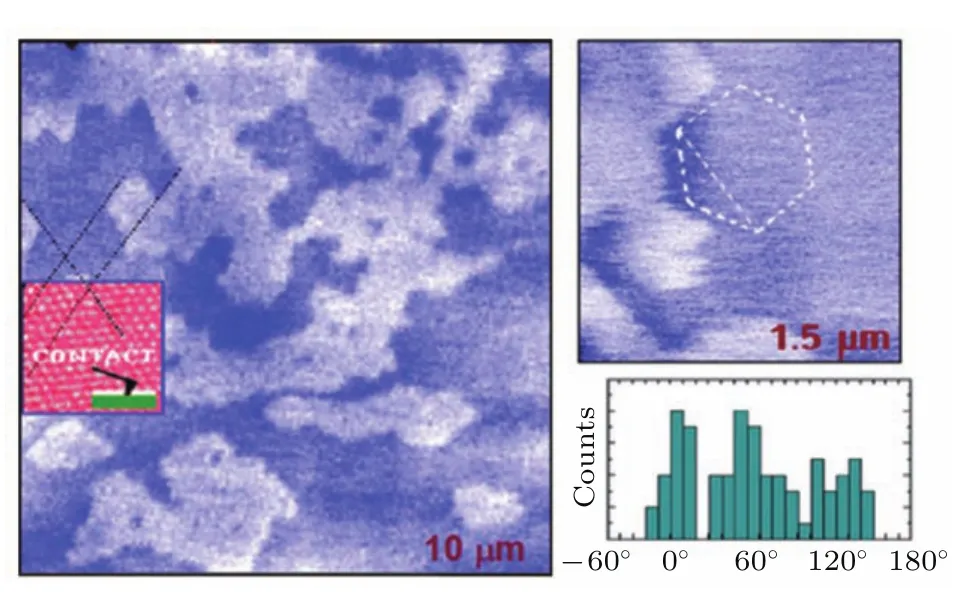
图18 云母上水结构的SPFM图 亮的的区域对应着第二层水,暗的区域为第一层水;边界处趋向于五元环,较小图案处为六边形状,它的方向和云母晶格相关(摘自文献[89])Fig.18.SPFM images of structures formed by water on mica.Bright areas correspond to a second water layer and dark areas to the first water layer.The boundaries tend to have polygonal shapes,as shown in the smaller image where a hexagon is drawn for visual reference.The directions are shown in the smaller image where a hexagon is drawn for visual reference.The directions are strongly correlated with the mica lattice(Images are from Ref.[89]).
2010年,Xu等[91]利用石墨烯覆盖在云母片上的水层上,研究了水吸附层的吸附高度.结果表明,第一层和第二层水的吸附高度为0.37 nm左右,这和单层冰的高度相同,得到室温下吸附在云母片上的第一、二水层为冰结构的结论.石墨烯的作用是固定水的吸附层,为在室温下探测表面水创造了条件.如图19通常在接近0◦时冰的层间距为c/2=0.369 nm,这个和观察到的大约0.37 nm的结果相近.并且图19(f)中箭头所指的方向指明第一层水岛的边界大约为120◦,这证明了在云母上第一层水也有冰的性质,是对前述利用SPFM研究水工作的进一步发展.
2010年,Kimura等[92]利用AFM的FM模式原位研究了云母表面的特定晶体位点的水结构.他们利用力谱逐点成像的方法,得到了针尖与表面相互作用力的二维分布.图20(a)和图20(b)分别是云母表面结构模型和AFM图.图20(c)是FM模式测得的表面水结构.从图中可以看出,在水和云母的边界处,水分子渗透到了云母六元环的中心;同时可以从图中区分出云母表面的三层水的位置.但是水结构在表面上不是相同的,如图20(c)所示,橙色椭圆处作者认为是云母表面吸附的钾离子或者钾离子的水合物.这种不均匀性来源于表面电荷分布的不同,而且第三层水受表面结构的影响要比第一、二层水小.同年,Fukuma等[93]利用3D-SFM(three-dimensional scanning force microscopy)的FM模式得到了云母上六边形上吸附水分子和横向水层的三维分布,研究表明靠近水环境的云母表面存在表面弛豫.2013年,Herruzo等[94]将这种三维的思路扩展到双模AFM上,它采用在两个频率下同时激励微悬臂的方式.这样不仅增加了二维方向的分辨率,而且可观测性大大提高.同时,他们还对Fukuma的工作进一步发展,能够在10 pN,2 Å和40 s的分辨率下对云母上水层进行成像.图20(d)是云母-水的三维图,左图显示在靠近云母时,观测值的振荡周期与水层宽度一致;右图上界面上的微扰可以分析得到云母上的原子结构.这些研究显示了NC-AFM在研究水结构的重要作用,极大地推动了水科学的发展.
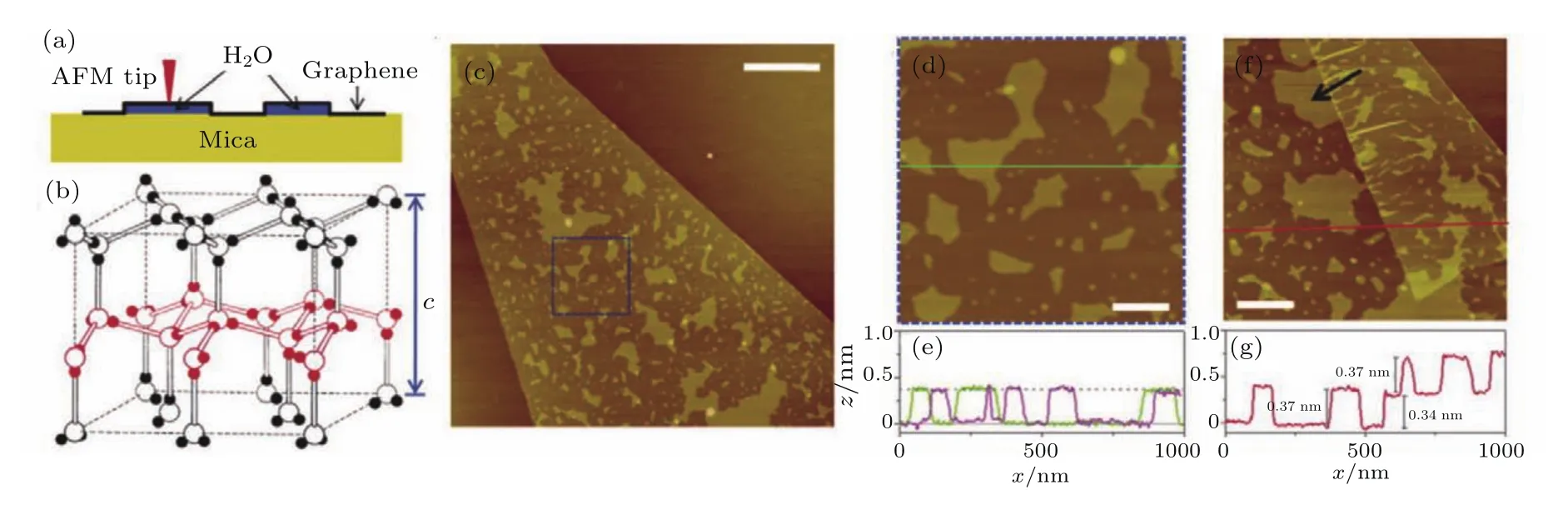
图19 在自然环境下利用石墨烯观察云母上第一层水 (a)AFM针尖探测石墨烯覆盖的水;(b)冰的结构;(c)云母上单层石墨烯的AFM图;(d)为(c)图中蓝色方块区域;(e)为横线处(绿色)和另一样品(紫色)的的高度信息;(f)另一样品的AFM图,单层石墨烯片的边缘发生了折叠;(g)图为横线处(红色)的高度信息(摘自文献[91])Fig.19.Graphene visualizes the first water adlayer on mica surface at ambient conditions:(a)A schematic of how graphene locks the first water adlayer on mica;(b)the structure of ordinary ice;(c)AFM image of a monolayer graphene sheet deposited on mica at ambient conditions;(d)a close-up of the blue square in(c);(e)height pro files along the green line in(d)and from a different sample,the dashed line indicates z=0.37 nm;(f)AFM image of another sample,where the edge of a monolayer graphene sheet is folded underneath itself,the arrow points to an island with multiple 120◦corners;(g)the height pro file along the red line in(f),crossing the folded region(Images are from Ref.[91]).

图20 (a)云母(001)表面结构的图示和(b)浸在水中的云母的AFM图;(c)二维水合结构力谱成像和图例,图中橙色椭圆处可能是吸附在表面的钾离子或者钾离子水合物;(d)水-云母界面的三维图,两图分别显示了水层结构和云母的原子结构 (摘自文献[92,94])Fig.20.(a)Schematic illustration of muscovite mica(001)surface structure;(b)FM-AFM image of muscovite mica in water,hexagonal structures are clearly observed;(c)2D hydration structure measured by FM-AFM and schematic illustration,the contrast is likely to be a K+ion or a hydrated K+ion complex adsorbed on the surface;(d)3D map of a mica-water interface,the side view shows the stripes are associated with the presence of hydration layers,right image illustrates the compatibility between 3D imaging and angstrom resolution(Images are from Refs.[92,94]).
室温下人们利用NC-AFM的许多模式来对表面水层进行成像和表征.随着低温技术的发展,水科学的研究也进入了新的领域,如在原子尺度上获得表面冰或者水的本征结构和动力学等.2013年,北京大学的Peng等[95]利用qPlus NC-AFM在液氦温度下研究了NaCl(001)表面上水团簇的高分辨成像,并且研究了成像的机理.实验和理论模拟比较后,关键在于在较大的针尖样品距离处探测CO针尖和极性水分子之间的高阶静电力.图21(a)是基于qPlus技术的NC-AFM,它的针尖尖端修饰了CO分子.图21(b)是在NaCl(001)表面吸附的水分子四聚体.四聚体具有两种不同的构型,水分子之间由氢键相互连接,每个水分子既是氢键电子的给体,也是电子的受体,自由的O—H键朝上远离样品表面.图21(c)和图21(d)是恒高度模式下的频率偏移图.当针尖高度较大时,四聚体呈现四个“耳朵”状;针尖高度变近后,氢键的环显现;距离更近时,中心环的衬度发生了反转.同时,他们使用四极矩()的针尖和中性针尖等其他针尖模拟,如图21(d),使用四极距针尖得出的结论和事实比较符合.同时发现,针尖的硬度k对成像影响不大.
基于这种高阶静电力成像,可以清晰地分辨水分子中氧原子和氢原子的位置.此外,由于CO针尖和水分子之间的高阶静电力相当弱,可以在没有任何扰动的情况下对很多弱键合的水分子团簇及其亚稳态进行成像.2018年,该研究组研究了钠离子的不同数目的水合物在NaCl(001)表面上的迁移过程.他们发现,钠离子与三个水分子形成的水合物相对于其他水合物在表面上扩散的要快.计算表明,这种高的迁移率来源于这种水合物的亚稳态结构,其中水分子可以以较小的势垒旋转.同时从该水合物中去除或者增加一个水分子都要增加扩散的势垒.该研究从原子分子尺度上分析表面水合物的扩散行为,为进一步理解表面水合离子传输提供了思路[48].
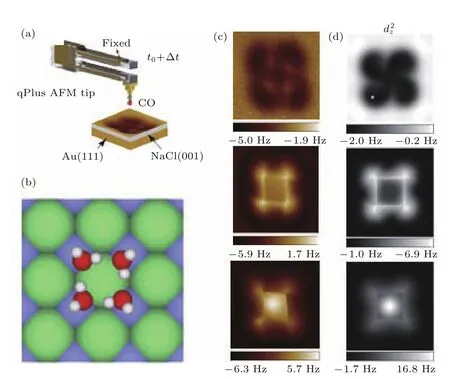
图21 (a)qPlus NC-AFM的图示(CO修饰的针尖);(b)NaCl(001)上吸附水四聚体的顶视图,氢、氧、氯、钠原子分别为白色、红色、绿色和紫色球;(c)针尖不同高度下的频率偏移图(100 pm,10 pm,-40 pm);(d)不同高度下模拟的AFM图像 (z1=7.9 Å,z2=6.8 Å,z3=6.4 Å)(摘自文献[95])Fig.21.(a)Schematic of a qPlus-based NC-AFM with a CO-tip;(b)top view of the water tetramer adsorbed on the NaCl(001)surface.H,O,Cl,and Na atoms are denoted as white,red,green,and purple spheres,respectively;(c)experimental∆f images recorded at the tip heights of 100 pm,10 pm,-40 pm,respectively;(d)simulated AFM images of a water tetramer with tip models,the images acquired at the tip heights of about z1=7.9 Å,z2=6.8 Å and z3=6.4 Å,respectively(Images are from Ref.[95]).
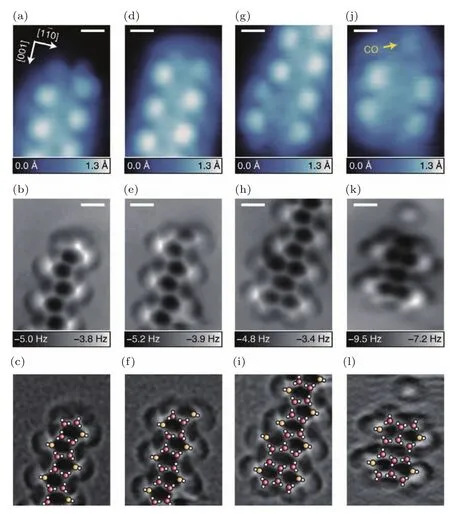
图22 水链终端的高分辨图像,五元环水链终止端STM(上)和AFM(中)图,原子结构图像(拉普拉斯滤波后)(下) (a)—(c)五元环终止;(d)—(f)五元环终止端有一个额外的垂直的水分子;(g)—(i)五元环和六元环融合;(j)—(l)四个五元环组成的团簇;标尺为5 Å(图片摘自文献[96])Fig.22.High-resolution images of terminals in water chains.STM(top)and AFM(middle)images of terminals for the pentagonal water chains.An atomic structure of each terminal is superposed on the Laplacian- filtered AFM image(bottom).(a)–(c)Pentagonal terminal.(d)–(f)Pentagonal terminal with an additional vertical H2O.(g)–(i)Fused hexagonal and pentagonal terminal.(j)–(l) ‘Tetraphyllous cluster’consisting of four pentagons.Scale bar is 5 Å(Images are from Ref.[96]).
由于在表面浸润过程中水层的缺陷具有重要的影响,2017年,Shiotari等[96]同样在液氦温度下利用qPlus NC-AFM和CO功能化的针尖研究了水网络的缺陷等问题.图22是一维水链三种终止端的高分辨AFM图像.除此之外,他们还研究了水-羟基网络和氢键的重排,从而对水网络在原子尺度上的缺陷进行了实空间的表征,为进一步理解界面水的结构提供了进一步的实验证据.
NC-AFM的快速发展为水科学的研究带来了新的机遇,同时也促进了该技术的发展.总之,在原子分子层面上操纵水以及理解水在界面处的行为,将会对水科学的发展提供新的契机.
3 总结与展望
从20世纪90年代初,扫描探针技术就开始用于界面(尤其是金属表面)水的研究,并且取得了丰硕的成果,这大大加深了人们对于界面水的结构和性质的认识,并对表面浸润水层的传统双层冰模型提出了挑战.近些年,人们逐渐将扫描探针技术扩展到更为复杂的绝缘体或者半导体表面和原位电化学环境,揭示出界面水很多新奇的物理和化学现象.在技术上,扫描探针技术的空间分辨率已经从单个水分子水平逐渐推向了亚分子级水平,使得在实空间和能量空间获取氢核的自由度成为可能,进一步推动了表面水的微观研究.
迄今为止,大部分高分辨扫描探针技术研究的界面水体系还比较简单和理想化,主要集中在表面上的水团簇、亚单层和单层水,如何将扫描探针技术应用于多层水和真实固液界面的研究将是未来的一个重要课题.最近发展起来的亚分子级分辨AFM成像技术显示出了很好的潜力,有望能在这个方向上发挥重要的作用.同时,我们也意识到,表(界)面水的研究还面临着很多问题和挑战,需要发展全新的扫描探针技术.
3.1 氮缺陷-空位(NV)色心技术
扫描探针显微学(scanning probe microscopy,SPM)的一个固有的问题就是会不可避免地扰动水的结构,不论是隧穿电子还是分子-针尖的相互作用力,这都是一个不可回避的问题.此外,高分辨的STM/AFM测量通常都会需要超高真空的背景环境和低温的工作条件,否则就不能够稳定成像.这些缺点使得SPM远不及传统谱学在水科学研究中应用的广泛,例如光谱、中子散射、核磁共振技术.近期,一种新型的利用NV作为探针的SPM技术提供了一种理想的在室温大气下非侵扰式成像的可能.
自然存在于钻石中的NV色心由于其原子级别的尺寸和靠近钻石表面(<10 nm),常常被用作纳米尺度的磁力计.它的孤对电子基态是自旋平行的三重态,但是可以通过激光极化并且通过自旋依赖的荧光读出.长的相干时间(0.1—1 ms)使得这个固态的量子探测器在大气环境下稳定并且容易被微波序列串相干地调控[97].未知的目标自旋或者是磁场都可以通过磁偶极相互作用被NV探测到.这使得对于类似于水中质子的自旋涨落这样极其微弱的信号可以在5—20 nm的范围内被探测到[98,99].同时,激发或者是读取的激光光束的功率是在几十毫瓦这样一个低的量级,加热的效应可以忽略不计.因此,NV-SPM是最有可能实现对于水结构非侵扰成像的工具.
除此以外,NV-SPM可以和低温超高真空兼容[100],也可以在大气和溶液环境下工作.包裹NV色心的钻石又是非常惰性的,可以适用于各种恶劣的环境,并且相干时间在一个大的温度范围(4—300 K)内变化很小[101].这是一个合适的连接超高真空和现实条件的实验技术.
最后,NV-SPM可以在纳米尺度进行核磁共振.这意味着精细的调节微波脉冲序列赋予NVSPM一系列的高分辨率(∼10 kHz)、高带宽(直流到∼3 GHz)的谱学的能力.这样就为探测到单个水分子内的质子磁共振信号提供了一种可能[102,103].
3.2 超快STM
STM仪器的电子学带宽通常被限制在MHz范围.但是氢键网络的动力学过程通常是在皮秒、或者飞秒的量级.这些过程包括质子转移、氢键的形成和断裂、氢键结构中的能量弛豫.这样的时间尺度的差别导致STM仅仅能够探测到初态和末态,却不能给出中间态或者过渡态的信息.如果能够将STM和超快激光技术结合,就可以同时实现原子级别的空间成像和飞秒级别的时间分辨[104,105].具体的实验中,有时间延迟的两束带激光被聚焦到STM中的针尖-样品结上,进而先后激发样品表面的分子.如果分子被激发,会在隧穿电流中引起一个瞬态的改变.进一步,如被第一束激光激发的分子在未弛豫的状态下被第二束激光照射,第二束激光将不会引起电流的变化.改变不同的延迟时间,就可以得到平均隧穿电流的变化.这些电流变化是可以被STM电学系统捕捉到的,所以惟一限制时间分辨的是激光脉冲的宽度.
激光结合的STM已经成功地被运用到半导体表面,主要集中于研究载流子动力学以及自旋弛豫动力学.如果要运用到水分子体系,需要进一步提高信噪比,并确保水分子在光照下的稳定.除此之外,激光对于STM针尖的热扰动是一个棘手的问题.在过去的10年内,许多研究组提出了不同的限制或者消除激光的热效应[106,107].综上所述,激光结合的STM技术是一个用来研究表(界)面的氢键体系动力学的极有利的工具.在不久的将来,可以预见激光STM会成为单分子层面研究超快动力学的强有力手段,并将改变许多对于水-固体界面的认识.
3.3 高压技术
水的相图非常复杂,在不同的温度和压强下可以得到不同的相.很多的晶体结构是在低温和高压下的亚稳态.低温下的结构中,通常氢键都是有序排列的,随着压强的增大,水分子通过弯曲氢键,形成紧密的环型或者是螺旋形的网络,最终能够得到更高密度的结构[108].但是缺乏一种能够局域对于水分子施加压力并能够原位表征的手段.上文中具有超高分辨成像能力的NC-AFM,如果牺牲部分分辨率,切换成接触模式(contact mode),就可以实现对于局域强压下水分子行为的表征,局域氢键网络结构的变化就能够反映在力谱中,也就是说能够看到随着压强的变化悬臂的受力存在突变或者不连续.另一方面,改变压强变化的速率,也许能得到平衡态以外的一些非平衡条件下的亚稳态.最重要的是,水分子结构的改变对应于能量的变化,力谱中可能会存在能量耗散导致的迟滞效应.利用原子力显微镜的空间分辨,可以在水和固体界面的不同位置进行力谱的探测,进而能够给出特征位置水分子的结构变化,从而帮助分析和理解水和界面之间的相互作用.
如果考虑到水分子间的氢键结构比较弱,难以承受很高的压强,可以利用一些特殊结构将水分子约束在其中,例如一维受限的石墨烯覆盖的水[91,109],二维受限的碳纳米管中的水[110,111],三维受限的绿宝石中的水[112].这些结构一定程度上限制了水分子的流动,从而能够实现对于少数水分子体系进行系统的研究.此外,受限体系中的水分子和界面的接触更多,此时,强压下的分子构型强烈依赖于水和界面相互作用.以上两点都可能导致不同以往的亚稳相出现,而它们的观测和表征对于水分子的本身及其与环境相互作用都是极为重要的.
仅仅利用力对于水分子的结构进行表征显得有些不足,可以通过在AFM悬臂上镀上金属实现对于水分子结构的电学表征.利用这种方法可以验证Pt(111)表面多层水的铁电性[113].与水分子化学结构相似的硫化氢被证实在高压下超导[114],这种高压诱导的结构和电学性质的变化是否会发生在水分子上引起了人们极大的兴趣.导电悬臂无疑给AFM的测量提供了更宽的维度,可以对水分子强压下奇异的电学性质进行系统的测量.
猜你喜欢
河北师范大学学报(自然科学版)(2022年5期)2022-09-20
波谱学杂志(2021年3期)2021-09-07
科教新报(2021年11期)2021-05-12
扬子江(2020年4期)2020-08-04
电子制作(2019年14期)2019-08-20
意林原创版(2017年11期)2017-12-01
少年文艺·开心阅读作文(2017年7期)2017-08-05
滇池(2017年5期)2017-05-19
中学化学(2015年12期)2016-01-19
中学化学(2015年12期)2016-01-19
