
金黄色葡萄球菌新型肠毒素SEK 原核表达、纯化及溶液构象分析
2019-03-11 08:44田万帆赵燕英唐俊妮
食品科学 2019年4期
田万帆,刘 骥,赵燕英,唐俊妮*
(西南民族大学生命科学与技术学院,四川 成都 610041)
由金黄色葡萄球菌(Staphylococcus aureus)分泌的肠毒素以其极端的热、酸、蛋白酶水解抗性[1-7]和干燥稳定性使之在食品加工的酸热处理[8]和食品保藏的过程中,仍然保持结构完整性,是导致形成食品中毒的重要毒力因子[9-10]。此外,由于具有直接结合抗原提呈细胞表面MHC II分子和T细胞表面T细胞受体,并能非特异性促进多克隆T淋巴细胞增殖能力,肠毒素也被称为超抗原[11]。
金黄色葡萄球菌K型肠毒素(S. aureus enterotoxin K,SEK)编码基因sek最早于2001年通过对S. aureus基因组序列比对分析而发现[12]。Jarraud等[13]研究表明,sek基因与编码肠毒素SEG、SEI、SEL和SEM的基因形成一个操纵子位于肠毒素基因簇。流行病学调查显示[14-15],sek不但是临床分离的致病性金葡菌菌株中最高频出现基因之一,更是与耐甲氧西林金黄色葡萄球菌(methicillinresistant S. aureus,MRSA)的mecA基因具有显著相关的肠毒素编码基因,这暗示含有sek基因的金黄色葡萄球菌在逃避宿主免疫系统攻击和抗生素抗性获得上可能发挥重要功能。因此,对于sek基因表达调控或对其表达产物SEK蛋白的靶向性抑制,有望成为临床上控制MRSA菌株的新策略。SEK蛋白归属于V型超抗原[16],与其他各型超抗原的不同之处在于,V型超抗原具有一个被称为α3-β8环的15 个氨基酸残基形成的插入序列,该序列位于第3个α-螺旋和第8个β-折叠之间。研究表明SEK蛋白的α3-β8环与T淋巴细胞表面T细胞受体的Vβ结构域相互作用时发挥关键作用[17],因而该插入序列可能成为抑制SEK超抗原活性的靶点。利用恒河猴[18]和臭鼩[19]进行的动物实验结果表明,SEK蛋白没有经典肠毒素SEA或SEB的显著催吐活性,但发生催吐的时间与经典肠毒素SEA或SEB无显著差异,说明SEK也是一种潜在的食品中毒致病因子,对其进行灭活研究是提高食品安全的重要手段之一。
尽管关于SEK催吐活性[18-19]、超抗原活性[12,17]、时序表达[20-21]、高灵敏度检测[22]以及单抗中和治疗策略[23]等方面被广泛研究,但迄今为止,关于SEK蛋白溶液构象的报道并不多见。众所周知,蛋白质生物学功能发挥依赖于天然蛋白形成正确的折叠态构象,因此,蛋白构象及其稳定性分析对于蛋白功能研究至关重要。鉴于SEK肠毒素引起食物中毒的普遍性和高发性,为进一步理解肠毒素SEK结构与功能关系,本研究通过优化SEK原核表达体系获得重组蛋白,进而利用荧光发射谱、圆二色谱以及十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDSPAGE)对SEK溶液构象进行研究,旨在揭示维持SEK溶液构象稳定性的分子基础,为探索破坏SEK结构稳定性,建立更加高效合理的食品消毒工艺奠定理论基础。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株与质粒
金黄色葡萄球菌SA005(该菌株携带enterotoxin K-like protein(selk)基因,其NCBI登录号为KU574280.1)保存于本实验室;感受态大肠杆菌(Escherichia coli)菌株DH5α、BL21(DE3)、BL21(DE3)pLysS、Rosetta(DE3) 天根生化科技(北京)有限公司;原核表达质粒pET-28a(+) 美国Novagen公司。
1.1.2 试剂
聚合酶链式反应(polymerase chain raction,PCR)扩增试剂、核酸分子质量标准、限制性内切酶EcoRI、SalI 宝生物工程(大连)公司;蛋白Marker 天根生化科技(北京)有限公司;SDS-PAGE 5×上样缓冲液康为世纪生物科技有限公司;牛血清白蛋白、胰蛋白胨、酵母粉 英国Oxoid公司;卡那霉素、氯霉素、异丙基硫代半乳糖苷(isopropylthiogalactoside,IPTG) 美国Sigma公司;thrombin 北京博奥拓达科技有限公司;丙烯酰胺、甲叉丙烯酰胺、咪唑、三羟甲基氨基甲烷(trismetyl aminomethane,Tris) 美国Amresco公司;Ni2+-NTA-Sepharose、DEAE Sepharose Fast Flow 美国GE Healthcare公司;10 kDa超滤管 德国Millipore公司。
含SDS和巯基乙醇SDS-PAGE上样缓冲液:10 mL含0.6 mL 1 mol/L Tris-HCl(pH 6.8)、5 mL 50%甘油、2 mL 10% SDS、0.5 mL β-巯基乙醇、1 mL 1%溴酚蓝、0.9 mL蒸馏水;含SDS不含巯基乙醇SDS-PAGE上样缓冲液:10 mL含0.6 mL 1 mol/L Tris-HCl(pH 6.8)、5 mL 50%甘油、2 mL 10% SDS、1 mL 1%溴酚蓝、1.4 mL 蒸馏水;Buffer A:500 mmol/L NaCl、50 mmol/L Tris(pH 8.0);Buffer B:1 mmol/L pH 7.0磷酸缓冲液;thrombin储液:1 000 U溶于1 mL磷酸缓冲液,储液浓度1 U/μL;其他试剂均为分析纯。
1.2 仪器与设备
HZQ-F160全温振荡培养箱 苏州培英实验设备有限公司;5804R型冷冻离心机 德国Eppendorf公司;WD800B型微波炉 格兰仕集团有限公司;TSNENEN031445 PCR仪、DYY-6C型核酸电泳仪、Mini-PROTEAN Tetra Cell电泳仪 美国Bio-Rad公司;JY92-IIN型超声破碎仪 宁波新芝生物技术有限公司;Chirascan plus圆二色谱仪 英国Applied Photophysics Limited公司;F-7000荧光光谱仪 日本Hitachi公司。
1.3 方法
1.3.1 ΔNspSEK蛋白原核表达载体的构建
根据上述模糊判断的结果,通过加权平均法就可以得到最终的精确输出量,即在特定偏移条件下,支撑油缸应采取的液压缸压力差。
引物以及ΔNspSEK(ΔN表示N端缺失,sp为信号肽signal peptide的缩写)蛋白原核表达载体构建参考文献[24],金黄色葡萄球菌基因组DNA采用本实验室建立的微波加热法提取[25],利用不含信号肽的sek基因上游引物序列:5’-CGTACGGAATTCCAAGGTGATATAGGA ATTG-3’(下划线为EcoRI酶切位点),下游引物序列:5’-CGCTAGGTCGACTTATATCGTTTCTTTATAAG-3’(下划线为SalI酶切位点,加粗体为插入的终止密码子)进行PCR扩增[24]。扩增所得片段经琼脂糖凝胶电泳检测及PCR产物纯化试剂盒纯化后,与质粒载体pMD18-T进行T-A连接,分别将插入ΔNspsek基因的pMD18-T质粒与空载质粒pET-28a(+)用EcoRI/SalI双酶切,将上述双酶切获得的基因片段用T4 DNA连接酶连接,使得目的基因定向克隆至表达载体。将插入ΔNspsek基因的质粒命名为pET-28a(+)-ΔNspsek,转化至大肠杆菌DH5α扩增,将提取到的质粒pET-28a(+)-ΔNspsek进行EcoRI/SalI双酶切鉴定。同时将提取到的质粒送成都擎科生物技术有限公司测序。
1.3.2 重组质粒在大肠杆菌中表达条件优化
将经过测序验证的重组质粒pET-28a(+)-ΔNspsek分别转化感受态大肠杆菌BL21(DE3)、BL21(DE3)pLysS和Rosetta(DE3)筛选高表达宿主菌株,在相应的抗生素抗性平板上(卡那霉素抗性质量浓度30 µg/mL;氯霉素抗性质量浓度34 µg/mL)随机挑取6 个含有重组质粒的单克隆菌落,分别过夜培养后,按1∶50的比例扩大至含相应抗生素抗性的LB培养基中,37 ℃振荡培养3 h,然后加入适当浓度诱导剂IPTG后,继续恒温振荡培养一定时间后,离心收集菌体,SDS-PAGE检测蛋白表达。
选择高表达菌株,分别进行诱导时间梯度(37 ℃,0.5 mmol/L IPTG分别诱导0、1、2、4、6、8、10、12、24 h),诱导IPTG浓度梯度(37 ℃,8 h,IPTG浓度梯度为0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8 mmol/L),诱导温度梯度(8 h,IPTG浓度0.5 mmol/L,诱导温度分别为17、27、37 ℃),以及Zn2+浓度梯度(0、1、5、10、15、20、25 mmol/L)进行优化,确定最佳诱导条件。
用获得的最佳条件进行小量诱导,12 000 r/min、10 min离心收集菌体,缓冲液(50 mmol/L Tris-HCl,500 mmol/L NaCl,pH 8.0)洗涤菌体2~3 次,重悬菌体进行超声破碎,超声破碎条件:功率30%,模式1,变幅杆Φ06,超声破碎3 s,停3 s,共5 min。随后12 000 r/min,离心10 min,收集上清液,沉淀以相同步骤再处理2 次,合并上清液为蛋白粗提液,蛋白粗提液与沉淀分别进行SDS-PAGE检测,目的蛋白大部分存在于上清液,表明蛋白为可溶性表达;目的蛋白大部分存在于沉淀则为包涵体表达。
1.3.3 His-ΔNspSEK蛋白分离与纯化
根据1.3.2节确定的最优表达菌株为出发菌种接种于5 mL LB液体培养基中,37 ℃过夜振荡培养。次日将种子液转接到500 mL LB中按照确定的最佳表达条件扩大培养,监测菌液吸光度A600nm至0.6~0.7时,进行IPTG诱导表达。离心收集菌体,菌体经PBS洗涤后重悬浮于20 mL Buffer A缓冲液中,冰浴超声破碎,超声破碎条件:功率285 W,振幅30%,模式1,变幅杆Φ06,超声破碎3 s,停3 s,共15 min,离心收集上清液。将得到的His-ΔNspSEK蛋白粗提液上样于事先用含Buffer A缓冲液平衡好镍离子亲合层析柱。先以Buffer A缓冲液和含50 mmol/L咪唑的Buffer A缓冲液除去大部分杂蛋白,再以含100 mmol/L咪唑的Buffer A洗脱目的蛋白,SDS-PAGE分析纯度。随后,采用凝血酶去除SEK His标签,取纯化的蛋白于截留分子质量10 kDa容量15 mL的Millipore超滤管脱盐至PBS(pH 7.4)中,作为凝血酶的酶切底物,配制1 IU/μL的thrombin储液。0.5、1.0、1.5 IU thrombin酶切300 μg SEK蛋白22 ℃、4 h。SDS-PAGE检测酶切是否完全。最后,将酶切后的蛋白更换为Buffer B缓冲液,采用阴离子交换去除凝血酶,获得纯化的ΔNspSEK蛋白。
1.3.4 ΔNspSEK重组蛋白聚合态和热稳定分析
将纯化的ΔNspSEK蛋白分别加入2 种不同的SDSPAGE上样缓冲液(一种是含SDS和不含巯基乙醇;另一种是同时含SDS和巯基乙醇)处理后进行SDS-PAGE分析,纯化的ΔNspSEK蛋白置于100 ℃水浴0、3、6、10、20、40、60 min,牛血清白蛋白作对照,SDS-PAGE分析蛋白的受热降解程度。
将纯化的ΔNspSEK重组蛋白用Buffer B调节质量浓度为0.1 mg/mL,参考文献[26]条件,在Chriascan plus型圆二色谱仪上进行蛋白溶液的测定,仪器参数设定为:扫描范围190~260 nm,激发光和发射光狭缝1 nm,中速扫描,光谱校正开启,25 ℃氮气吹扫下进行。每个样品扫描3 次取平均值。参考文献[26]条件将纯化的ΔNspSEK蛋白调节质量浓度为45 μg/mL,干式恒温器25 ℃温育10 min,后用F-7000荧光光谱仪进行测定,仪器参数设定:激发波长278 nm和295 nm,发射波谱扫描范围为300~400 nm,扫描速率为1 200 nm/min,扫描激发光和发射光狭缝为5 nm,PMT电压700 V,消除光栅,每个样品扫描3 次取平均值。
1.4 数据分析和图像处理
使用圆二色谱数据在线分析软件DichroWeb对扫描的圆二色谱数据进行分析,选择CDSSTR拟合方案,采用圆二色谱参考数据集SMP180,190~240 nm波长范围内对扫描数据进行拟合,计算溶液中ΔNspSEK蛋白二级结构含量。将得到荧光光谱数据用FL Solutions转化成Excel表格,然后用Origin7.5作出278 nm和295 nm波长处荧光光谱图。
2 结果与分析
2.1 重组表达质粒pET-28a(+)-ΔNspsek的基因测序鉴定结果
将重组表达质粒的插入基因ΔNspsek进行测序,得到长度为729 bp的片段,利用NCBI BLAST多序列比对发现克隆到的ΔNspsek基因序列与菌株SA005的selk基因序列(登录号:KU574280.1)的第70~729位碱基序列100%吻合,第1~69位为信号肽序列。质粒双酶切鉴定结果见图1,表明SEK融合蛋白(His-ΔNspSEK)表达载体构建成功。
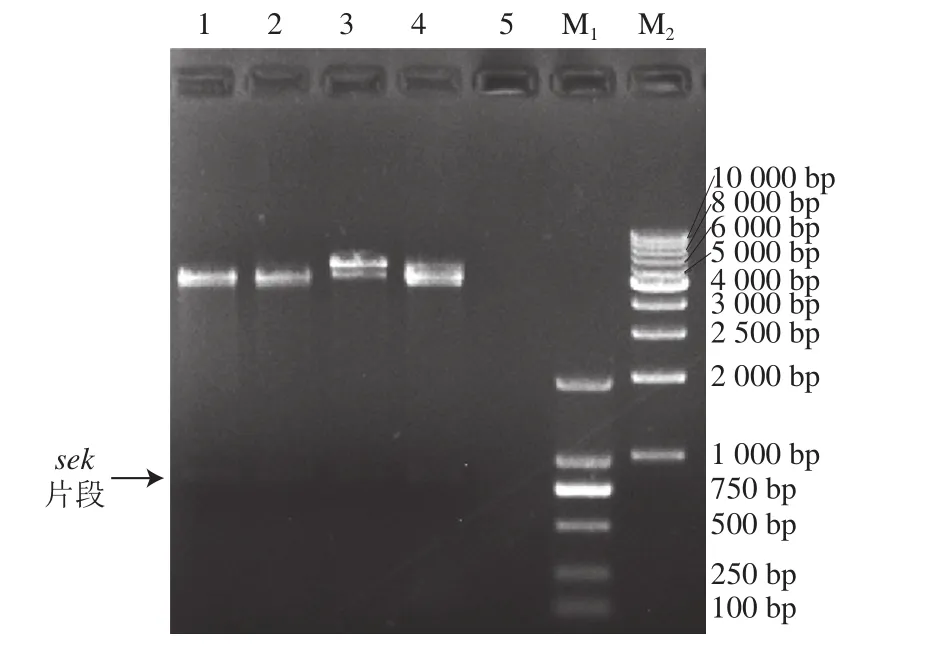
图1 重组质粒pET-28a()-ΔNspsek的双酶切鉴定Fig. 1 Veri fication of pET-28a(+)-ΔNspsek positive clone by doublerestriction enzyme digestion
2.2 pET-28a(+)-ΔNspsek在大肠杆菌中表达条件优化及可溶性表达验证结果
将重组原核表达质粒pET-28a(+)-ΔNspsek分别转化至BL21(DE3)、BL21(DE3)pLysS、Rosetta(DE3)3 种不同的感受态细胞,对每个转化子在平板上随机挑选6 个单克隆,经过相同的条件进行小量诱导表达,结果如图2所示。目标蛋白在3 种不同的宿主菌中均有明显表达,且Rosetta(DE3)表达宿主菌中蛋白产量普遍较高(图2C),该菌株整合了大肠杆菌缺乏的6 种稀有密码子对应的tRNA,因此,选取表达量最高的Rosetta(DE3)菌株中的1号单克隆作为后续表达条件优化实验的菌株,进一步探讨融合蛋白的最佳表达条件。
由图2D可知,当IPTG浓度梯度升高至0.6 mmol/L时,目标蛋白表达量最大,随着IPTG浓度升高,杂蛋白也在增加,为减轻纯化负担,本研究选择0.5 mmol/L为最佳IPTG浓度。由图2E可知,随着诱导时间的延长,目标蛋白表达量也在逐渐增加,但8 h后增加不明显,因此,选择最佳诱导时间为8 h。由图2F可以看出,低温诱导目标蛋白表达量较大,因此,设置最佳诱导温度为27 ℃。由图2G可知,低浓度Zn2+并未显著改善目标蛋白表达量,而当Zn2+浓度高于10 mmol/L时,目的蛋白几乎不表达,因此,Zn2+浓度选择为0 mmol/L。综上所述,选择IPTG浓度0.5 mmol/L、诱导时间8 h、诱导温度27 ℃、不添加Zn2+为最佳诱导条件。
利用上述最优表达条件对Rosetta(DE3)宿主菌1号单克隆进行小量诱导表达验证,由图2H可知,3 次平行实验均获得ΔNspSEK蛋白高产量表达。随后,将1 mL菌液离心收集菌体,经Buffer A缓冲液重悬后超声破碎,裂解液离心所得上清液见图2I的泳道1,离心的沉淀见图2I的泳道2,上清液中有清晰的蛋白表达条带,结果表明融合蛋白为可溶性表达。
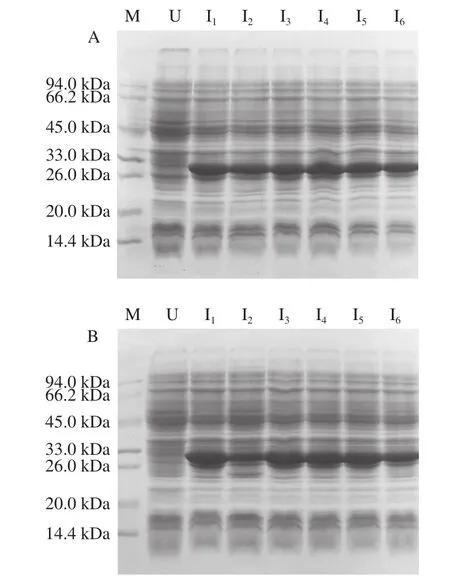

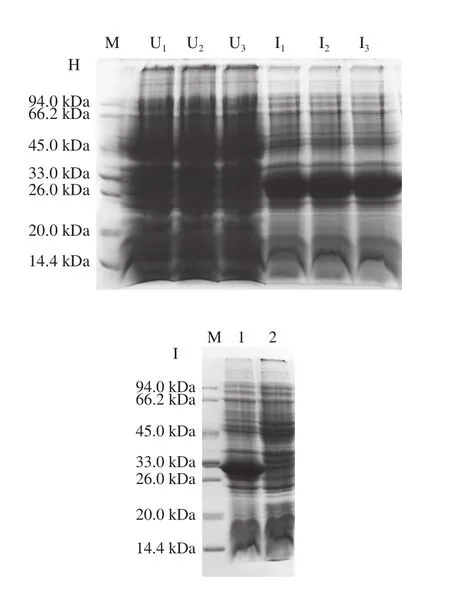
图2 ΔNspsek基因在不同原核表达宿主菌中诱导表达及条件优化Fig. 2 Optimization of expression conditions for His-ΔNspSEK protein
2.3 最优条件下的His-ΔNspSEK表达纯化及凝血酶切除6×His标签结果
按上述最优条件扩大培养所得融合蛋白超声破碎粗提液进行镍亲合层析纯化,经过含10、50 mmol/L咪唑的缓冲液梯度洗脱杂蛋白后,含His标签的ΔNspSEK融合蛋白被100 mmol/L咪唑缓冲液洗脱效果好,见图3A的第5泳道。进一步采用凝血酶切除6×His标签肽,图3B的泳道3效果较好,对于300 μg融合蛋白底物,凝血酶量为1.0 IU时,酶切4 h即可完全除去标签肽。进一步将凝血酶切除标签肽后的反应体系超滤去除标签肽并更换为Buffer B,收集过DEAE离子交换树脂的穿透液进行SDS-PAGE,结果如图3C中的泳道2所示,结果表明除去His标签肽的ΔNspSEK蛋白纯度高,可以满足后续光谱学测定。
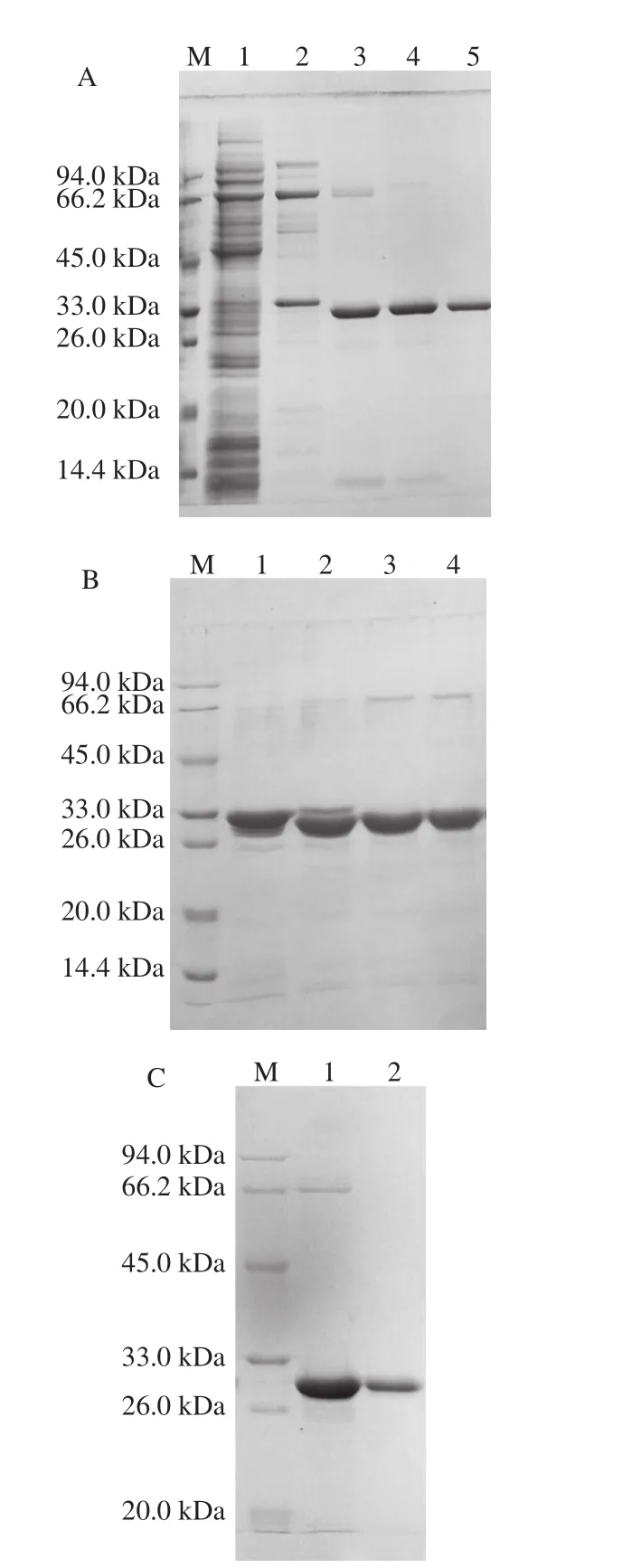
图3 His-ΔNspSEK蛋白的纯化与凝血酶切除6×His标签肽Fig. 3 SDS-PAGE analysis of purified His-ΔNspSEK fusion protein after thrombin digestion
2.4 纯化的ΔNspSEK聚合状态分析和热稳定分析
将凝血酶切除标签后经DEAE纯化的ΔNspSEK蛋白分成2 份,分别用含有SDS与β-巯基乙醇的样品处理液(泳道1)和只含有SDS不含β-巯基乙醇的样品处理液(泳道2)处理后,进行SDS-PAGE,结果如图4A所示,由于SDS可以破坏蛋白质亚基间的非共价相互作用,β-巯基乙醇可破坏链内或链间二硫键,因此,泳道1显示的是ΔNspSEK蛋白单体形式。泳道2中样品由于缺乏β-巯基乙醇,不能破坏链间二硫键,部分ΔNspSEK蛋白以二聚体形式呈现。结果表明,在非还原态时,截除信号肽的SEK可能通过链间二硫键形成二聚体的形式存在。进一步将去除His标签肽的ΔNspSEK重组蛋白100 ℃热处理,间隔时间为0、3、6、10、20、40、60 min进行采样,分别对样品进行SDS-PAGE分析,如图4B所示,100 ℃加热到40 min时,重组蛋白ΔNspSEK依旧无明显降解,但到加热60 min时可以看出蛋白出现部分降解,说明ΔNspSEK蛋白具有较高的热稳定性。
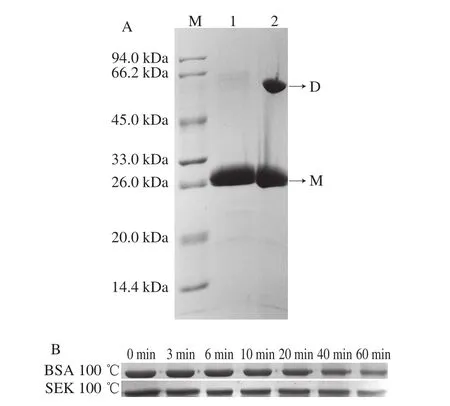
图4 ΔNspSEK蛋白聚合状态分析(A)和热稳定性分析(B)Fig. 4 SDS-PAGE analysis of aggregation state (A) and heat stability (B) of ΔNspSEK
2.5 纯化ΔNspSEK蛋白的圆二色谱和荧光发射谱分析
纯化的重组蛋白ΔNspSEK远紫外区的圆二色谱见图5A,表现为192 nm波长处的强正峰和208 nm波长处的强负峰信号。采用DichroWeb在线分析软件,计算溶液中ΔNspSEK重组蛋白二级结构含量为α-螺旋29.1%、β-折叠23.6%、β-转角28%和无规卷曲19.4%,Difference(exp-recon)表明实验数据与拟合数据之间的误差在可接受范围之内。由图5B可知,蛋白溶液分别在278 nm和295 nm波长激发时,具有相同的344 nm波长色氨酸荧光发射峰。与处于亲水环境的游离色氨酸354 nm波长荧光发射峰相比蓝移了10 nm,说明虽然ΔNspSEK蛋白中色氨酸处于蛋白质分子表面,但仍未完全暴露于极性水环境中。
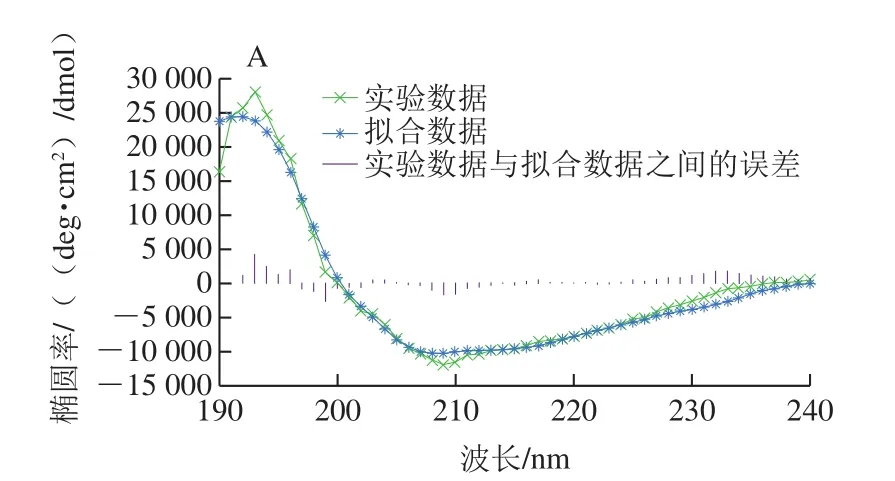
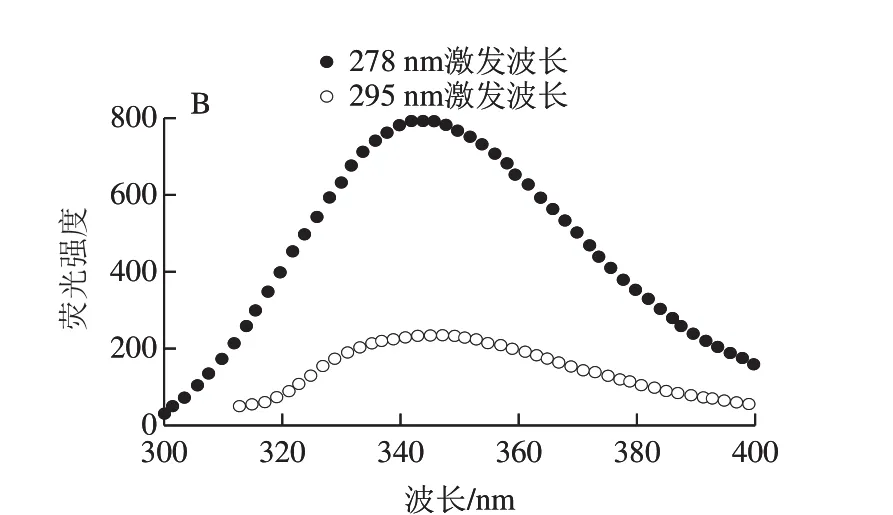
图5 ΔNspSEK重组蛋白的远紫外圆二色谱(A)与室温荧光发射谱(B)Fig. 5 Far-UV circular dichroism spectra (A) and room temperature fl uorescence emission spectra (B) of ΔNspSEK fusion protein
3 讨 论
本研究对SEK蛋白表达条件进行优化,并建立表达ΔNspSEK蛋白的方法。在纯化蛋白前通过查找文献得知ΔNspSEK理论等电点为7.7。而采用凝血酶酶切后杂蛋白的等电点为5.0~5.5[26-27],据此,本研究尝试使用阴离子交换方法进行蛋白质酶切纯化。通过多次实验证明当缓冲液pH值为7.0时,穿透后,凝血酶结合到了DEAE柱上,ΔNspSEK蛋白大部分保留在穿透液中,这样的尝试大大降低了酶切后的纯化难度,此方法可供相关研究者借鉴。
众所周知,加热对于食品加工与安全具有重要作用,是食品杀菌的主要手段。本研究对金黄色葡萄球菌肠毒素重组蛋白ΔNspSEK的热稳定进行研究,通过圆二色谱分析揭示ΔNspSEK蛋白溶液构象β-折叠为23.6%。刘骥等[26]研究认为新型肠毒素M的蛋白溶液构象β-折叠高达42%,王小红[28]研究表明传统B型肠毒素β-折叠达40.4%,经过121 ℃处理30 min后,仍具有39.9%的β-折叠含量。β-折叠对肠毒素热稳定性维持具有关键作用,SEK的β-折叠含量比SEM和SEB的都低,因此,可以解释SEK的热稳定性不如SEM和SEB好,当100 ℃加热到60 min时,蛋白出现部分降解。如果在食品加工中延长加热时间可以破坏部分SEK肠毒素,但是还会有部分SEK稳定存在,需进一步探索破坏SEK的方法。
对融合蛋白ΔNspSEK的荧光发射谱进行分析,发现278 nm波长激发时呈现的是色氨酸特征性的344 nm波长荧光发射主峰。该峰位与295 nm波长单独激发色氨酸残基时,产生的荧光发射峰一致,根据蛋白质分子内能量转移有效性和色氨酸荧光峰位相对于极性水环境(354 nm波长发射峰)的显著蓝移,可以推测纯化的ΔNspSEK处于构象紧密的天然折叠状态。
本研究成功构建SEK的融合蛋白ΔNspSEK可溶性原核表达系统,纯化的融合蛋白ΔNspSEK在溶液中处于构象紧密的天然折叠状态,ΔNspSEK蛋白热稳定性好,为进一步研究SEK的结构与功能关系提供参考依据。
猜你喜欢
猪业科学(2021年3期)2021-05-21
数学大王·低年级(2020年8期)2020-08-14
生物工程学报(2019年1期)2019-01-30
中国医药指南(2019年14期)2019-01-07
天然产物研究与开发(2018年10期)2018-11-06
农家科技下旬刊(2017年3期)2017-04-26
小雪花·初中高分作文(2016年9期)2016-05-14
中国医药生物技术(2015年4期)2015-12-26
分析化学(2014年7期)2014-12-13
食品工业科技(2014年5期)2014-03-11
