
水合铀酰与尿嘧啶及其异构体配位体系的计算研究
2019-03-13 03:06牟永晓曹建平翁约约王朝杰
无机化学学报 2019年3期
牟永晓 曹建平 翁约约,2 孙 祥,3 卫 涛 王朝杰*,
(1温州医科大学药学院,温州 325035)
(2温州市中心医院药剂科,温州 325000)
(3温州医科大学附属第一医院药学部,温州 325000)
0 引 言
随着军事和核能工业的快速发展,如何处理核废料以及如何避免核污染一直是国际社会共同关注的难题。有数据显示,现阶段全世界443座运行的核反应堆已累积了3.6×105吨致命的高放射性核废料,并且以每年1.2×104吨的速度增长[1]。传统核燃料主要为锕系元素中的铀,是一种放射性损伤核素,半衰期长,造成的核污染可持续数十万年以上,而且其具有的化学毒性常被轻视。铀的核外层电子组态为5f36d17s2,自然条件下以六价态形成的铀酰离子(UO22+)最为稳定[2-3]。UO22+可与多种无机或有机配体形成配合物并逐渐渗入地下水和土壤中[4],最终不可避免地与生物体接触,对人类和环境具有潜在的危害性。Garmash[5]等通过体外研究发现,低浓度的铀酰离子会造成DNA和蛋白质结构的氧化损伤,其毒性主要与铀酰离子物理化学性质有关。具有高灵敏度、高亲和力的铀特异性脱氧核酶,已广泛应用于环境中铀酰离子的生物监测[6]。因此研究铀酰离子与多种生物有机小分子的结合行为有助于我们更深入地了解铀对生物体的影响。除了实验研究外,理论计算也是研究铀酰离子与生物小分子相互作用的重要途径,可为实验研究铀酰离子在生物机体内的吸收、转运、沉积的分子机制提供理论支持。
目前国内外对铀酰离子和有机小分子的理论研究已有大量报道。如Zakharieva等[7]应用相对论密度泛函方法研究溶液中铀酰离子与含氮有机小分子配位形成[UO2(H2O)4(LN)]2+(LN=NH2CH3、N(CH3)3、NC5H5)的相互作用性质,并与五水合铀酰离子作比较,结果显示甲胺配合物最稳定,吡啶配合物的能量与五水合铀酰离子相差不大,而三甲胺配合物的能量比五水合铀酰离子高约40 kJ·mol-1。Wu等[8]报道了铀酰离子与丝氨酸和磷酸化丝氨酸在溶液中的配位结构和能量学性质,研究发现体系更倾向于形成五配位铀酰配合物,且当铀酰离子与配体以1∶3配位时配合物的结合能最高。Ren等[9]也通过密度泛函理论方法,研究了在水溶液中水合铀酰离子与氧代二乙酸、亚氨基二乙酸、硫代二乙酸形成饱和配位时的配合物结构特性和电子结构。Vukovic[10]等对水合UO22+与胺肟阴离子配合物进行了理论计算,其中胺肟阴离子中氮和氧在铀酰的赤道面上发生双齿配位,并将计算结果与实验测定UO22+与胺肟及其衍生物阴离子配合物的结果进行了比较。与大多数研究UO22+离子与氨基酸或多肽分子配位特征的理论研究相比,UO22+离子与核酸碱基分子或DNA片段相互作用的体系仍然较少[11-12]。
核酸的生物学行为及生物活性都是以构成其基本单元的碱基分子的性质为基础,而尿嘧啶(Uracil,Ur)是十分重要的核酸碱基分子,其6种互变异构体的结构和光谱性质已有大量研究报道[13-18]。铀酰离子介导的光裂解可用于RNA结构的构象分析,如Wittberger等[19]研究发现RNA二级结构的单链区域是铀酰的裂解位点。在tRNA中,铀酰裂解位点是在环结构内部以及中心铰链区。
本文应用密度泛函理论方法在相对论有效势基组水平对水合铀酰离子与尿嘧啶及其异构体形成的一系列配离子的结构与性质进行了计算研究,为进一步研究(脱氧)核苷酸或(脱氧)核糖核酸片段与锕系酰离子的配位行为和机制提供基础。
1 计算方法
迄今对于含锕系元素体系的计算报道甚多[20-23],以全电子(all-electon,AE)的相对论计算最为认可,但与可用的实验数据如振动光谱和结构参数比较,也并未体现出绝对优势[24]而且效率受限,其他模型势(model potential,MP)或赝势(pseudopotential,PP)的有效核势(effective core potentials,ECPs)及其基组得到更多的应用[25],但其计算结果常常与计算体系、计算方法尤其是密度泛函方法的选取有相关[26-29],本工作运用大核赝势和最广泛使用的杂化泛函来探索。
选用杂化密度泛函B3LYP方法[30],此泛函是研究铀酰离子配合物体系常用的计算方法之一[31-35],且表明对锕系酰离子的基态结构描述较为合理。计算中对铀原子采用Stuttgart RLC ECP[36]赝势基组,是包含了相对论效应的有效势和价层DZ基组,其中有效势对铀内层电子 1s~5s、2p~5p、3d~5d、4f共78个电子做冻芯处理,外层的14个电子(5f36s26p66d17s2)作为价层电子,其他原子采用6-311++G(d,p)基组。 对[UO2(Uracil)j(H2O)k]2+(Uijk,i=1~6,j=1~5,k=5-j,i为6种尿嘧啶异构体代号,j表示尿嘧啶配位数,k表示水分子配位数)体系的几何结构参数、振动光谱和能量学等性质进行计算,振动光谱分析均没有虚频,可确认其为稳定结构。
分子中的原子理论(Quantum theory of atoms in molecules,QTAIM)用于分析体系中配位作用和电荷转移变化。根据Bridgeman等[37]研究结果,在计算Mayer键级和QTAIM分析时对其他原子宜采用6-311G(d,p)基组。采用极化连续介质模型(polarized continuum model,PCM)[38]模拟水溶液对配离子的溶剂化效应。所有计算工作使用Gaussian09[39]程序包结合Multiwfn3.5[40]程序完成。
2 结果与讨论
2.1 水合铀酰尿嘧啶配离子的结构性质
首先我们对6种尿嘧啶异构体作了B3LYP/6-311++G(d,p)水平的优化计算,其几何结构参数和相对能量数据列于支持信息图S1和表S1,按气相相对吉布斯自由能由低到高的顺序依次编号为Ur1~Ur6,6种异构体均含有羰基和(或)羟基基团且只连接在C2和C4位上,其中Ur1属于双酮式,Ur4为双羟基结构,其余4种异构体之间的差别在于羟基和羰基的连接位置不同(如Ur2与Ur6、Ur3与Ur5)。最稳定的尿嘧啶(Ur1)比羰基-烯醇式异构体(Ur2)能量低48.4 kJ·mol-1,6种尿嘧啶异构体稳定性顺序与文献报道的计算结果一致[15-16]。
据相关铀酰离子配位体系的研究报道,当配体在铀酰轴的赤道面上与铀原子形成五配位键时可达饱和配位,且该饱和配合物在气相和水溶液中均比对应的不饱和配位结构稳定[41-44]。计算尿嘧啶Ur1与水合铀酰五配位的结构(U1jk),得到14种稳定结构(支持信息图S2)。从图S2可知,Ur1与铀酰离子的相互作用有2种类型,即环上的羰基氧(C2=O7和C4=O8)分别与铀原子配位形成U-O配位键。表S2列出了14种配合物的相对吉布斯自由能。结合图S2和表S2,当Ur1以C4=O8参与配位时,配离子能量最低,与对应的以C2=O7键参与配位的结构相比,两者能差在34.7~88.9 kJ·mol-1范围内。 而配位键类型相同,配体间空间位置不同的结构能量十分接近,后文中我们只对比分析最稳定的配合物结构。
五水合铀酰离子及U1jk体系在气相和水相中计算得到的U=O键长值接近于实验[45]测定值(0.170 2 nm)。文献中运用相同方法下计算其它体系得到的U-OH2键长值在0.252 0~0.265 6 nm范围[46-47],与我们结果一致。Ur1羰基氧与铀原子的配位距离为0.230 1~0.239 9 nm,与实验[48]测得的其它酮类结构(如 N,N,N′,N′-四正丁基丙二酰胺)中的羰基氧与UO22+的配位距离(0.236 0 nm)相近。

图1 五水合铀酰离子和水合铀酰尿嘧啶配离子[UO2(Uracil)jH2O)5-j]2+(j=1~5)在气相中的平衡结构Fig.1 Equilibrium structures of uranyl pentahydrate and hydrated uracil coordination uranyl ions[UO2(Uracil)j(H2O)5-j]2+in gaseous phase
随着Ur1配位数目的连续增加,配离子中U=O键、U-OH2键和U-Ouracil键均有不同程度的伸长,说明配体与中心铀原子间的作用整体上呈变弱的趋势。例如U=O键长从五水合铀酰离子的0.172 4 nm增长至U150配离子的0.176 0 nm,对应的Mayer键级则从2.35逐渐降低至2.14。另外U-Ouracil键长从 0.230 1 nm(U114)增长至 0.241 9 nm(U150),对应的键级则从0.53降至0.39,U-OH2键长从五水合铀酰离子的0.252 3 nm增长至U141体系中的0.258 7 nm,键级则从0.31减弱为0.26。U-Ouracil键长均比U-OH2键短,反映出尿嘧啶与铀酰离子的作用强度大于水分子与铀酰离子的作用强度。
考虑溶剂化效应后,在水溶液中U=O键和UOuracil键长伸长,而U-OH2键长缩短,在文献中也同样发现溶剂化后U=O键和U-Oligand键减弱,但U与水分子的作用增强[42,49],可能由于水环境会使得水的电子分布被极化,导致水中氧的孤对电子效应增加,加强了与铀原子的静电作用。在Ur1配位数为1~2的结构中,U-Ouracil键增长明显,平均增长了0.034 5 nm。而在U141和U150结构中,水溶液与气相中的U-Ouracil键长接近。表明在高配位数下,溶剂对铀酰尿嘧啶的配位影响逐渐减小。
一分子的尿嘧啶异构体与水合铀酰离子作用形成的饱和Ui14结构如图2所示。尿嘧啶异构体与中心铀原子存在3种配位类型:(1)羰基O与U的单配位, 如结构 U114、U214、U314、U514, 其中Ur1、Ur2、Ur3的环平面与铀酰轴平行,Ur5的环平面与铀酰轴垂直;(2)羟基O的单配位,如结构U414;(3)羰基O和环上N的双齿配位,如结构U613,由于饱和配位数是5,水分子配体数为3。Ui14体系的相对自由能数据列于支持信息表S3。从表S3可知,在气相中的相对能由低到高顺序为U514<U314<U114<U214<U414,因 U613 水分子配体数目不同,故不将U613结构纳入相对能量的比较。在结构U514与U314中,Ur5、Ur3环上的N均与邻位的水分子形成了N…H-O氢键,彼此气相吉布斯自由能量相差仅为1.5 kJ·mol-1,而U214的相对能差高达 37.3 kJ·mol-1。

图2 水合铀酰离子与6种尿嘧啶异构体在气相中形成1∶1型饱和配离子的平衡结构Fig.2 Equilibrium saturated structures of 1∶1 type complexes of hydrated uranyl ion with six kinds of uracil tautomers in gaseous phase
Ui14体系的几何结构参数列于表1中,表中数据表明U=O键的键长在各体系中较为接近。U-Ouracil键的键长递增顺序为 U214(0.229 9 nm)<U114(0.230 1 nm)<U514(0.230 5 nm)<U314(0.234 1 nm)<U613(0.243 5 nm)<U414(0.250 4 nm),对应的键级大小顺序则相反,而U-OH2键距按U613(0.251 6 nm)<U414(0.252 5 nm)<U314(0.253 9 nm)<U514(0.254 5 nm)<U114(0.255 6 nm)<U214(0.255 6 nm)的顺序递增。可见U-Ouracil键长与U-OH2键长整体上呈相反变化,表明尿嘧啶异构体与水分子配体存在明显配位竞争作用。Ui14结构U-OH2的平均键级为0.29,与文献[39]报道的四水合过氧铀酰[UO2(O2)(H2O)4]2+中U-OH2的键级(0.30)相接近。值得指出的是,U414结构U-Ouracil的键级为0.28,显著低于其它5种结构,且它与U514的气相吉布斯自由能相差高达182.9 kJ·mol-1,同时在水溶液环境的优化计算中,U414结构无法正常收敛,可见在水溶液中不能稳定存在。通过对相对能量和键级的分析,表明异构体Ur4与铀酰离子的作用强度较弱,由于Ur4是以羟基氧配位。考虑溶剂化效应后,各结构U-Ouracil键和U-OH2键的键长递增顺序较气相下发生改变。

表1 在气相和水相中水合铀酰尿嘧啶及其异构体配离子的结构参数和Mayer键级Table 1 Equilibrium structure parameters(nm)and Mayer bond orders of hydrated uracil and its tautomers coordination uranyl ions in gaseous and aqueous phases
2.2 水合铀酰尿嘧啶及其异构体配离子的振动光谱
U1jk结构中主要键的振动频率计算结果列于表2。由于B3LYP方法的基频校正因子都接近于1[50],因此不考虑基频校正因子。从表2中可知,五水合铀酰离子中O=U=O键的振动频率为对称(νss)961 cm-1和反对称(νas)1 039 cm-1,高于文献中[49]通过B3LYP/6-31+G**(RECP)水平计算得到的O=U=O键对称(937 cm-1)和反对称(1 026 cm-1)伸缩振动频率。水溶液中O=U=O键的伸缩振动频率与实验测定值[51](νss870 cm-1,νas965 cm-1)相差在 67 cm-1范围内。
在水溶液中,当Ur1分子逐步取代水分子后,O=U=O键对称和反对称伸缩振动频率逐渐降低,表明配位体系稳定性增加[52],辜家芳等[47]的报道也证实了同样的结论。因此,结合前文几何结构分析,说明尿嘧啶分子比水分子更易于与铀酰离子结合。与自由状态下Ur1环上氨基键(N-H)的伸缩振动频率3 616 cm-1相比(表S4),配位结构中的Ur1环NH键发生红移,且红移范围在45~50 cm-1间,说明羰基氧的配位削弱了邻位的N-H键。
计算的铀酰尿嘧啶异构体配离子(Ui14)相关键的振动频率数据也列于表2中,配位的O=U=O伸缩振动频率较为接近(除了U414结构)。6种结构的O=U=O键对称伸缩振动频率按照U214(935 cm-1)<U514(938 cm-1)<U114(939 cm-1)<U314(941 cm-1)<U613(946 cm-1)<U414(949 cm-1)的顺序递增,对应结构的U-Ouracil键长整体也呈变长的趋势。U414结构O=U=O键的伸缩振动频率与[UO2(H2O)5]2+结构伸缩振动频率相接近,说明尿嘧啶环的羟基氧与铀酰离子的相互作用较弱,与前文几何结构分析结果一致。考虑溶剂效应后,发现水相结构中O=U=O键的伸缩振动频率红移,而νN-H蓝移。

表2 在气相和水溶液中水合铀酰尿嘧啶及其异构体配离子中UO22+的对称(νss)和反对称(νas)伸缩振动频率、尿嘧啶及其异构体环上N-H键和O-H键的伸缩振动频率Table 2 Vibrational frequencies(νssand νas)of U=O bond and vibrational frequencies of N-H and O-H bonds of uracil and its tautomers in hydrated uracil and its tautomers coordination uranyl ions in gaseous and aqueous phases
2.3 水合铀酰尿嘧啶及其异构体配离子的能量学分析

为了分析配离子稳定性及配体与铀酰离子的结合强度,计算了Uijk体系的总结合能及尿嘧啶和水配体分别与铀酰离子作用的单结合能,单结合能计算类似于“第一电离能”的计算方法,计算公式如下:将配位体系分为铀酰离子、尿嘧啶、水分子3个片段,其中EUi(j-1)k、EUij(k-1)分别表示失去一分子尿嘧啶和一分子水的配合物的能量,而Eib、Ewaterb表示尿嘧啶和水分子的单结合能,即在配离子中单个配体的结合能量。计算以上相互作用能时均考虑了基组重叠误差(basis set superposition error,BSSE)校正。 计算得到的各项结合能数据列于表3中。从表3可知,气相中[UO2(H2O)5]2+的结合能为-1038.0kJ·mol-1,Spencer等[53]在 BLYP/DZP(ECP)计算水平,不考虑BSSE校正得到[UO2(H2O)5]2+的结合能为-1 073.6 kJ·mol-1,与本文计算的结合能相一致。
在U1jk系列结构中,随着尿嘧啶配体数目的增加,总结合能逐渐增大,配体的单结合能在逐渐减小。U-Ouracil键的结合能比U-OH2键的结合能大,体现尿嘧啶与铀酰离子的作用比水分子与铀酰离子的作用强,但是与文献[47]中计算得到的铀酰酸根配 合 物[UO2(CO3)2]2-(-688 kJ·mol-1)和[UO2(NO3)3]-(-367.7 kJ·mol-1)的单结合能相比较,发现尿嘧啶的单结合能较和的小,这表明带负电荷的酸根阴离子比中性尿嘧啶分子更易与铀酰离子发生相互作用。在水溶液中,水分子的单结合能低于气相下的结合能,说明U-OH2配位键的键能降低,体现出溶剂效应。
为了定量描述结合能与Ur1配体数量(0~5)间的关系,我们使用Origin软件对U1jk体系的结和能进行拟合,拟合曲线如图3所示。由于在水溶液中配体单结合能的变化趋势不明显,故只对气相下的单结合能进行拟合分析。在气相下,得到尿嘧啶配位数(j)与总结和能(ETotalgas)的拟合结果为:
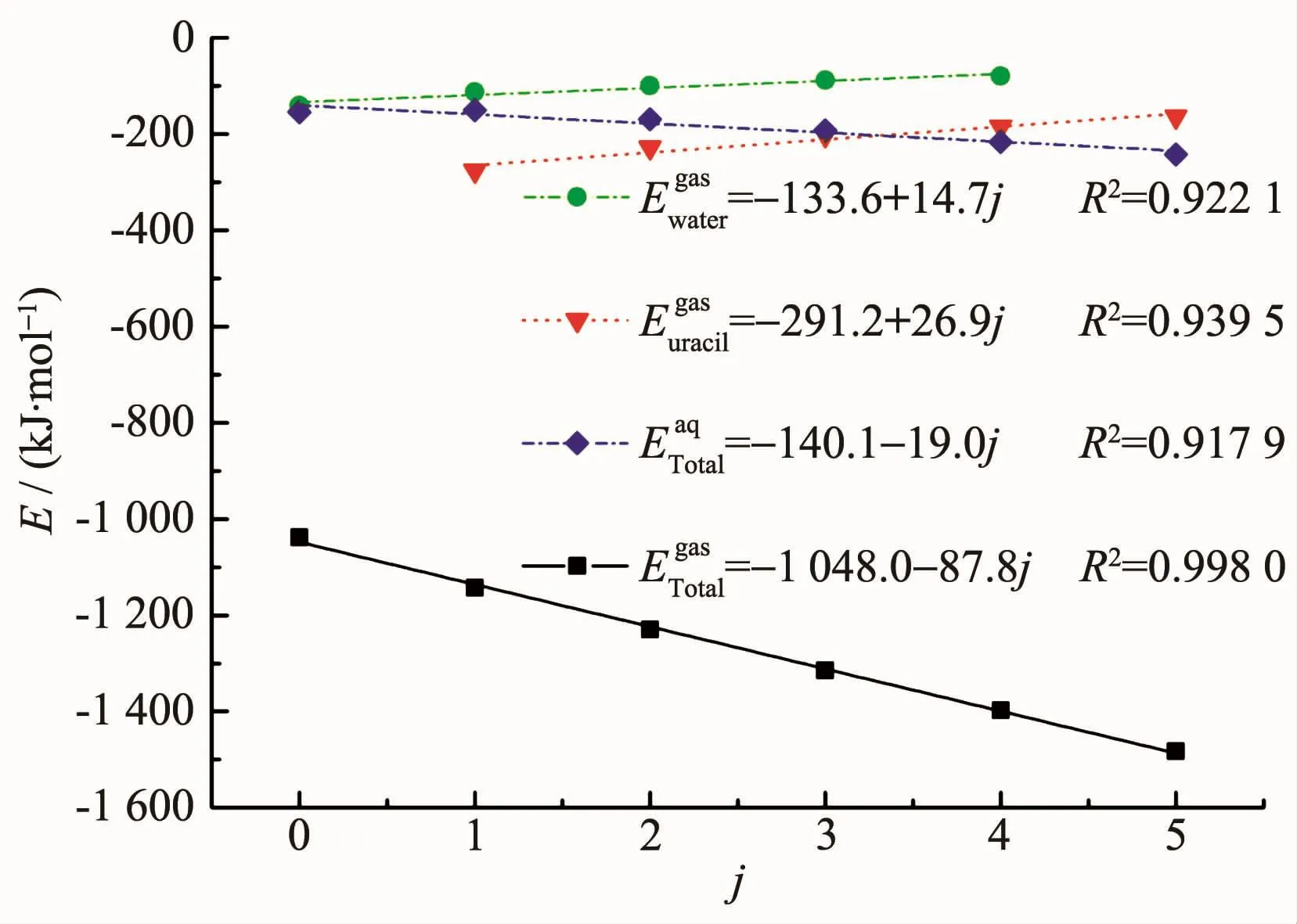
图3 U1jk体系在气相和水溶液中结合能与Ur1配位数j的关系Fig.3 Relationships between binding energies and the number j for Ur1 ligand of U1jk systems in gaseous and aqueous phases

在水溶液中,得到j与ETotalaq的拟合结果为:

可见气相下总结和能的拟合度R2值更接近1,表明气相下线性拟合的效果较好。水溶液中的总结和能增加量较小,为19.0 kJ·mol-1,这可能是由于Ur1分子极性较大,嘧啶环上的氨基H和羰基O均可与水分子形成氢键,故考虑溶剂化效应对结合能的变化影响较小。
更负的结合能表明配体与铀酰有更强的相互作用,体系亦更稳定。比较Ui14体系的总结合能在-1 014.1~-1 238.5 kJ·mol-1的范围,见表 3。 Ui14体系中结合最紧密的(U514)不是由能量最低的Ur1异构体生成,但是Ur5本身存在的概率较低。U514结构的总结合能在气相和水溶液中均最负,这与Ur5环上的N与相邻的水分子形成了配体间氢键相关,协同增强。U613结构中尿嘧啶的单结合能(-432.5 kJ·mol-1)最大,体现配体与铀酰离子配位的成键数目越多,两者的相互作用越强。U414结构中尿嘧啶的单结合能(-143.6 kJ·mol-1)与水分子的单结合能(-133.2 kJ·mol-1)略接近。由于溶剂效应的影响,水溶液中配体的单结合能显著减小。

表3 在气相和水溶液中水合铀酰尿嘧啶及其异构体配离子的结合能EBSSETable 3 Binding energy with the BSSE correction for hydrated uracil and its tautomers coordination uranyl ions in gaseous and aqueous phases
2.4 配离子的拓扑分析
为了进一步研究体系中配位键的形成和强度,我们使用Multiwfn程序进行QTAIM拓扑分析。根据 Bader[54]理论,当 BCP处的 ρ(r)大于 0.2 a.u.且▽2ρ(r)为负值时,说明该键具有共价键性质,若BCP处的 ρ(r)小于 0.2 a.u.且▽2ρ(r)为正值时,则该键具有离子键性质。表4列出了气相中配位键在(3,-1)临界点处的电子电荷密度和电子密度拉普拉斯值。从表 4中 U1jk体系的 U-Ouracil键在 BCP处的 ρ(r)和▽2ρ(r)分别在 0.058~0.072 a.u.和 0.236~0.326 a.u.范围内,U-OH2键在 BCP 处的 ρ(r)和▽2ρ(r)分别在 0.039~0.043 a.u.和 0.156~0.174 a.u.范围内,说明 U-Ouracil键和U-OH2键具有离子键的特征。文献[8]报道了水合铀酰离子与丝氨酸分子相互作用形成的U-Ocarboxyl配位键在 BCP 处的 ρ(r)约为 0.046,且▽2ρ(r)均为正值,同样具有离子键特征,与本文U-Ouracil键的QTAIM分析结果一致。U-Ouracil键和U-OH2键的ρ(r)和▽2ρ(r)数值整体上随着Ur1配体数目的增加而逐渐减小,表明其离子键特征在逐渐减弱,在Ur1配位数由 3 增加到 4 时,U-OH2键的 ρ(r)和▽2ρ(r)值反而略有增大,与上文在气相中对应的键长变化趋势一致。
Ui14体系中U-Ouracil配位键在BCP处的ρ(r)按照 U214(0.075 a.u.)>U514(0.073 a.u.)>U114(0.072 a.u.)>U314(0.068 a.u.)>U613(0.063 a.u.)>U414(0.050 a.u.)的顺序递减,U214、U514、U114 的 ρ(r)值相近,而 U-Ouracil键的 ρ(r)和▽2ρ(r)的大小顺序与前文的尿嘧啶异构体单结合能强弱顺序不同。6种配离子中U-OH2键的 ρ(r)范围在 0.043~0.047 a.u. 之间。Lipkowski等[55]提出了氢键的键临界点 ρ(r)和▽2ρ(r)的范围标准,分别在 0.002~0.040 a.u.和 0.020~0.150 a.u.之间。U514和U314结构中,尿嘧啶环上N与邻位水分子形成的N…H-O键在BCP处的ρ(r)和▽2ρ(r)在上述氢键范围内,可确认其形成了氢键。

表4 气相水合铀酰尿嘧啶及其异构体配离子配位键临界点处的电子密度(ρ(r))和拉普拉斯值(▽2ρ(r))Table 4 Electron density(ρ(r))and Laplacian of electron density(▽2ρ(r))at U-O(N)and H2O-N bondcritical point of hydrated uracil and its tautomers coordination uranyl ions in gaseous phase
2.5 电荷转移分析
配体间因相互作用而重新平衡后,不仅几何结构会发生改变,其原子上的电荷也会发生重新分布。为了进一步研究目标体系的电子结构性质,我们对相互作用的3个配体片段(铀酰离子、尿嘧啶及其异构体、水分子)进行了自然布居分析(natural population analysis,NPA),以此来考察在形成配离子前后各配体片段的电荷分布变化情况。
对于U1jk体系,表5列出了在气相和水溶液中形成配离子后3个配体片段所带的NPA电荷。与自由状态下的片段电荷相比,可以看出形成配离子前后各配体所带平均电荷变化明显,其中尿嘧啶和水分子片段所带平均电荷数目变大,如在气相中分别增加了0.307~0.383和0.185~0.193。而铀酰离子所带电荷数目从自由状态下的2.000降低至0.465~0.845范围内。从铀酰离子片段电荷的降低(得电子)和配体片段的电荷升高(失电子)可说明在配合过程中是由配体片段向铀酰离子发生了电子转移。同时,尿嘧啶向中心受体转移的电子数大于单个水分子向中心受体转移的电子数。配体向中心受体转移的电子数越多,形成的配位键越强。在U1jk体系中,随着Ur1配体数量的增加,铀酰离子所带电荷数从0.845逐渐减小至0.465,即得到的电子数逐渐增加(1.155至1.535),而尿嘧啶片段的平均电子转移数从0.383减少至0.307。考虑了溶剂效应的影响后,Ur1片段的电荷变化量略有下降,而水分子所带的电荷数略升高,此与前文在水环境下UOuracil键和U-OH2键的键长互有增减的结果相一致。表明溶剂效应会影响配离子中配体间的电荷重新分布。
对于Ui14体系,各配体片段所带的电荷列于表5中。铀酰离子所带电荷数按照U214(0.831)<U514(0.837)<U114(0.845)<U314(0.853)<U613(0.857)<U414(0.908)的顺序递增,表明Ur2异构体和水分子向中心铀酰的电子转移数最多。而U613结构中尿嘧啶异构体片段所带电荷数最高 (0.507),Ur6异构体失电子数最多,进一步表明该异构体与铀酰形成的双配位键会增加电子向中心片段的转移量。

表5 在气相和水溶液中水合铀酰尿嘧啶及其异构体配离子中各配体的自然布居(NPA)电荷数Table 5 Natural population analysis(NPA)for charges of each ligand in hydrated uracil and its tautomers coordination uranyl ions in gaseous and aqueous phases
3 结 论
采用B3LYP杂化密度泛函方法在相对论有效势基组和6-311++G(d,p)水平下对[UO2(Uracil)j(H2O)k]2+(j+k=5)配位体系进行研究。对几何结构、振动光谱、结合能、电子密度拓扑参数、原子电荷转移等进行了阐述,得到以下几点结论:(1)配离子结构表明配位键及U=O键的键长随Ur1配体数目的增加而伸长,且U-Ouracil键长较U-OH2键短。Ui14体系中U214的U-Ouracil键长最短。键长大小与Mayer键级成负相关关系;(2)振动光谱分析表明,在水溶液中O=U=O键的伸缩振动频率随Ur1配体数量的增加而减小,较气相下发生红移,而νN-H发生蓝移;(3)结合能计算表明配离子的总结合能随Ur1配体数目的增加而增加,但配体单结合能逐渐减小。Ui14结构中U514的结合能最大,并与结合能最低的U414结构相差232.6 kJ·mol-1,可见Ui14体系中结合最紧密的结构并不是由能量最低的Ur1异构体生成;(4)QTAIM拓扑分析表明配位键具有离子键性质,且临界点的 ρ(r)和▽2ρ(r)整体上随着 Ur1配体数目的增加而减小,键能降低,与配体的键长变化趋势一致;(5)原子电荷分析明确了在配合过程中是由配体片段向铀酰发生了电子转移,且尿嘧啶及其异构体所带电荷量大于单个水分子的电荷量,同时尿嘧啶的所带电荷量与配体数量大致成负相关关系。由于大核赝势在轨道作用分析方面存在不足,在后续锕系酰离子与其它碱基、碱基对、(脱氧)核苷酸或(脱氧)核糖核酸片段作用体系的探索中将对大小核赝势进行对比研究。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
腐植酸(2020年5期)2020-12-20
人工晶体学报(2019年5期)2019-06-18
当代陕西(2019年6期)2019-04-17
World Journal of Clinical Cases(2019年4期)2019-04-16
科技创新与品牌(2019年12期)2019-02-06
分析化学(2018年2期)2018-03-02
绿色科技(2017年20期)2017-11-10
科技创新导报(2017年19期)2017-09-13
外语学刊(2014年3期)2014-12-03
湖南师范大学学报·自然科学版(2014年4期)2014-10-23
