
共表达网络分析识别心肌分化过程中的关键因子
2019-04-26 05:03张艺
电子技术与软件工程 2019年4期
文/张艺
心脏疾病伴随着心肌细胞的坏死,已经坏死的心肌细胞会完全失去收缩功能,无法起死回生。心肌细胞再生能够使心脏得到修复,使受损的心脏得到功能的完善,直接重编程能够为这一目标的实现提供帮助。与干细胞技术相比,直接重编程把一类终末分化细胞直接转化为其他类终末细胞,而不用经历干细胞阶段。在很短的时间内就可以获得功能更佳的心肌细胞,诱导的时间也会很短。采用自体细胞不会受到伦理因素的限制,而且可能进行在体内直接转分化,不必进行细胞移植,能够避免形成肿瘤的风险、存活率低以及免疫排斥反应的种种问题。直接重编程诱导方向较为精确,也不会产生靶细胞之外的细胞类型。不过,心肌细胞直接重编程技术还不成熟,无法得到到成熟的心肌细胞,仅仅有一小部分细胞拥有自发电活动,自发收缩的功能;因为心肌细胞自身很难进行增殖,所以要获取大量数目的细胞用于临床是很困难的。
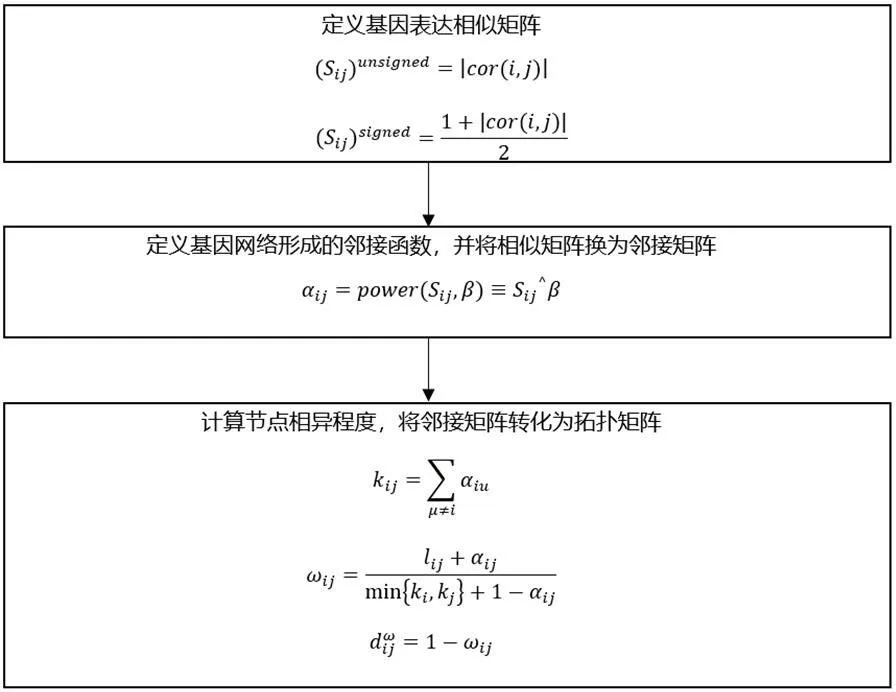
图1:WGCNA算法基本步骤

图2:共表达网络
Davis等[5]在1987年的发现奠定了细胞重编程的基础。2010年,Ieda等[6]受到多种转录因子组合诱导多能干细胞技术的启发,首次报道了通过过表达三个心脏发育的核心转录因子Gata4、Mef2C、Tbx5(GMT),可在体外成功诱导成纤维出现心肌样细胞表型。之后,Song等[7]在经典三因子的基础上加入Hand2(GHMT),可以使成年心脏成纤维细胞及鼠尾成纤维细胞,更有效地转化为心肌样细胞;Protze等[8]用胚胎成纤维细胞,筛选重编程因子,发现用Mef2C、Myocd、Tbx5(MMT)组合能够上调心肌细胞的表达;Wada等[9]在GMT的基础上,加入Mesp1和Myocd(GMTMM),能使人成纤维细胞表现出更多心肌细胞特点。整体来看,核心的重编程因子,能在一定程度上,把成纤维细胞转为心肌细胞的状态,但是效率并不高,如何高效地获取更多功能完备的心肌细胞成为一个难题。因此提高成纤维细胞直接重编程为心肌细胞的效率,需要更深入研究转录机制,寻找更高效率的转录因子组合。
单细胞基因组测序技术在近些年来发展很快。传统RNA测序方法,可单次处理大量的细胞,但得到的是平均表达水平。不存在两个完全相同的细胞。单细胞RNA测序(scRNA-seq)可以挖掘出单个细胞独具的基因表达变化。利用scRNA-seq技术,在单细胞的水平,分析心肌细胞发育的机制,会更有利于挖掘出重要的转录因子,重新对转录因子进行组合,从而提高重编程为心肌细胞的效率。

图3:心肌相关聚类模块
1 数据和方法
1.1 数 据
数据来自NCBI,数据库编号为GSE76118,是2016年Sean等[11]人的工作,采集了胚胎发育第8.5天(e8.5), e9.5和e10.5天的心脏单细胞转录组数据。
1.2 方法
1.2.1 数据处理
选取233个组织和细胞类型相关的候选核心转录因子,从下载好的RNA-Seq数据中筛选出这些候选转录因子的所有单细胞数据。过滤掉零表达和低表达的转录因子,最终剩余1258个转录因子作为研究对象。原始数据的基因表达值单位是CPM(count-per-million),在接下来的数据分析过程中需要进行对数化,取log2(CPM+1)。
1.2.2 共表达网络构建方法
构建共表达网络的步骤为,根据基因与基因之间的相关性关系判定两基因之间的权重大小。这个关系描述了基因表达模式之间的相似性。相关性的计算方法有斯皮尔曼相关系数、皮尔逊相关系数等等。每个节点代表一个基因,每条边代表共表达关系的强弱,利用相关性来构建出网络。相关系数值分布于-1到1之间。在无向网络中,取相关系数的绝对值。
大量的文献选择利用皮尔森相关系数来计算基因之间的权重,这样的做法存在一个明显的缺陷,阈值的取值通常是人为定义,取值过小容易造成网络体积过大,冗余信息干扰到关键基因的发掘,取值过大则容易丢失有用信息。为避免这样的处境,权重基因共表达网络分 析(Weighted Gene Co-Expression Network Analysis,WGCNA),利用软阈值的判定方法,能够对这个问题进行化解。
作为一种系统生物学分析方法,WGCNA构建了一个幂指数邻接函数,对相关系数取n次幂,来进行加权。用邻接系数αij=power(Sij,β)=|Sij|β,在加权系数β的作用下,相关系数的分布,趋向于无尺度分布。利用WGCNA分析方法构建加权共表达网络的步骤如图1所示。
1.2.3 从网络中寻找功能聚类模块
利用cytoScape中的插件MCODE(Molecular Complex Detection),在庞大的网络中进行聚类,构建功能模块。MCODE计算网络中各个节点信息。以该点为种子节点,能扩展出的最大k值k-core,此K-core的密度以及该节点的分数值。节点的分数反映出,这个节点及其周边的节点的密集度。之后,从分数值最大的节点开始,调用函数getClusterCore( ),利用该节点为种子节点,进行扩展,然后加入符合参数条件的邻接节点。最后,据参数的要求进行后续的处理,得到功能模块。
1.2.4 筛选网络中枢纽基因
虽然多细胞生物中的所有细胞基本共享相同的DNA序列,但在每一种细胞类型中,仅有一组特定的基因被主动表达。不同的细胞类型被不同的活性基因组合和它们之间的相互作用所管控。在每个细胞内,一组靶基因和调节基因,即转录因子(TF),彼此相互作用并形成基因调控网络。基因调控网络在拓扑上包括高度连接的组件和几个具有低连接性的节点。识别网络中的关键基因,能够更好的理解细胞命运的转变挑战在于如何找到掌控细胞命运转变的基因。枢纽基因根据网络拓扑性结合生物学意义来进行挑选。
对网络中的节点使用cytoHubba进行网络拓扑参数计算。cytoHubba包括节点连接度、边缘渗出组件、最大邻居组件、最大邻居组件的密度、最大团中心性、瓶颈值、偏心度、紧密度、发散性、中介性、应力11个节点参数的计算。结合生物学意义,选用以下几个参数来度量网络中转录因子的重要程度。
假定G=(V,E)是个无向图,V代表网络中的点集,E是边集。对于一个集合S,用|S|来表征它的基数(集合中的元素个数)。
a. 度(Degree,Deg):
Deg(v)=|N(v)|,节点的度越高,该节点相连的节点越多,代表其重要性越高。
b. 紧密度(Closeness, Clo):
c. 中介性(Betweenness,BC)
d. 瓶颈值(Bottleneck, BN)
Ts表示以s为根节点的最短路径树。BN(v)=∑s∈Vps(v),当多于|V(Ts)|/4条从s出发的路径在v点汇合,ps(v)=1;否则ps(v)=0。经过所有的瓶颈节点有一个共同特点,就是它们的邻居节点可分为多个不相交的节点集合,这些节点集合需要互相通信交流的时候,只能经过该瓶颈节点。故瓶颈值越大,重要性也越高。
2 结果

表1:7个枢纽基因
2.1 共表达网络构建
从预处理后的数据中筛选出1258个转录因子,并提取这些转录因子的表达矩阵,通过上述方法对网络进行构建,如图2。
2.2 鉴别共表达网络中心肌相关模块
通过cytoScape中MCODE工具的应用,在共表达网络中识别到了8个共表达模块,通过分析网络模块,图3所示的模块与心脏发育的过程紧密相关。
2.3 筛选枢纽基因
从图3模块中挑选枢纽基因,得到影响心肌发育的枢纽基因。
首先通过cytoHubba计算每个节点的Degree、Closeness、Betweenness、Bottleneck等参数值,评定出候选的18个枢纽基因。经过R语言的GOstats包对18个枢纽基因进行GO富集分析,最终综合筛选出Ctnnb1、Nme2、Nf ia、Gata6、Hmgb1、Hmga1、Notch1(如表1)这7个枢纽因子在心肌细胞生长过程中发挥着极其重要的作用。
3 讨论
通过对心肌发育的单细胞转录组数据进行预处理,利用加权基因共表达网络分析算法,对1258个转录因子构建共表达网络,通过MCODE聚类方法识别到心肌相关的模块,通过网络拓扑性的特征参数计算和GO富集分析最终筛选出重要的候选转录因子。其中,Notch1转录因子已被研究证明,对胚胎期心脏的发育有影响,并且和成年期动物心血管系统疾病的关系十分密切,Gata6转录因子对心肌能量代谢及细胞增殖具有重要作用。深入研究这些转录因子将是下一步工作的重要研究内容,应用生物学实验方法确认这些因子在重编程过程中的作用也是今后工作的重点。
总体来说,直接重编程和干细胞技术相较,跳过了干细胞的阶段,在短短的时间内可以获得功能更佳的心肌细胞,诱导时间大大减少。不过心脏直接重编程技术还面临着许多挑战,对心脏直接重编程分子机制的了解还不够深入。研究表明小鼠和人类的重编程过程也有所不同,如转录因子不同,需要的时间不同等。从目前的技术到转化为临床应用还是有很大距离,仍存在很多问题,需要利用更多新颖的计算机技术方法来进行持续深入的挖掘与探索。
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年7期)2021-11-04
少先队活动(2021年2期)2021-03-29
昆明医科大学学报(2020年12期)2021-01-26
学生天地(2020年17期)2020-08-25
数学大王·低年级(2020年3期)2020-03-12
山东医药(2017年20期)2017-07-01
海南医学(2016年8期)2016-06-08
中国学术期刊文摘(2016年1期)2016-02-13
中国病理生理杂志(2015年8期)2015-12-21
