
Loss of cavin1 and expression of p-caveolin-1 in pulmonary hypertension: Possible role in neointima formation
2019-05-17 00:46JingHuangRajammaMathew
World Journal of Hypertension 2019年2期
Jing Huang,Rajamma Mathew
Abstract
Key words: Caveolin-1; Cavin-1; Neointima; Phospho-caveolin1; Pulmonary hypertension
INTRODUCTION
Pulmonary hypertension (PH),a sequel of a number of unrelated systemic diseases,has a high morbidity and mortality rate.Regardless of the underlying disease,endothelial dysfunction and/or disruption plays a pivotal role in initiating vascular changes such as impaired vascular relaxation,and the activation of proliferative and anti-apoptotic pathways; subsequently leading to medial hypertrophy,elevated pulmonary artery pressure and right ventricular hypertrophy (RVH).Progressive pulmonary vascular changes lead to neointima formation resulting in the irreversibility of PH[1].Significant advances have been made in the field of PH since it was described by Ernst von Romberg in 1891.Modern therapy has improved the quality of life; however,the vascular changes continue unabated[2,3].One and 3-year survival rate for the patients with pulmonary arterial hypertension (PAH,Gr 1) is reported as 85%,and 58% respectively[4,5].The mechanism of PAH,especially the neointima formation is not yet understood.The situation is compounded by the fact that the diagnosis of PH is often made after significant pathological changes have already taken place in the pulmonary vasculature.
The experimental models have significantly contributed to the understanding of the pathogenesis of PH.We have previously shown that a single subcutaneous injection of monocrotaline (MCT) results in the disruption of endothelial cells (EC),loss of endothelial cavoelin-1 (cav-1) and reciprocal activation of proliferative and antiapoptotic pathways such as PY-STAT3,Bcl-xL,and increased proliferating cell nuclear antigen (PCNA) expression before the onset of PH.The endothelial disruption and endothelial cav-1 loss are progressive; and PH is observed at 2 wk post-MCT.At 4 wk post-MCT,85% arteries exhibited loss of endothelial cav-1; and a loss of von Willebrand factor (vWF) was observed in about 29% of pulmonary arteries.Loss of vWF was present only in the arteries with endothelial cav-1 loss.The loss of vWF,accompanied by the endothelial cav-1 loss,is indicative of extensive EC disruption and/or loss.At this stage,about 23% of the arteries,only with combined vWF and endothelial cav-1 loss exhibited enhanced expression of cav-1 in SMC[6,7].It is worthy of note,that the increased levels of circulating vWF[8],and EC[9],have been reported in patients with irreversible PAH.Importantly,loss of endothelial cav-1 associated with enhanced expression of cav-1 in smooth muscle cells (SMC) has been observed in PAH associated with congenital heart defect[10],drug toxicity[11]and in idiopathic and heritable PAH[12,13].Subjecting MCT-treated rats to hypobaric hypoxia accelerated the disease process; and by 4 wks,neointimal lesions were observed.Importantly,neointimal lesions were seen only in the arteries that exhibited extensive loss of endothelial cav-1 and enhanced expression of cav-1 in SMC[12].Furthermore,SMC from idiopathic PAH inin-vitrostudies have revealed enhanced expression of cav-1,increased capacitative Ca2+entry and DNA synthesis,which can be attenuated by silencing cav-1[13],indicating that this cav-1 in SMC has become pro-proliferative.
ECs form a semi-permeable monolayer between the circulating blood and the underlying tissue.Cav-1,a 22 kD membrane protein is localized in caveolae,flaskshaped lipid rafts found on the plasma membrane.Cav-1 is expressed in most cell types such as EC,SMC,epithelial cells,fibroblasts and adipocytes.Cav-1 interacts with transducing factors that are present in or are recruited to caveolae; and it maintains them in a negative conformation.However,cav-1 has been shown to have a pro-proliferative aspect depending on the disease state and the cell context[14].Interestingly,tyrosine 14,phosphorylated cav-1 (p-cav-1) is thought to inactivate growth inhibitory function of cav-1 scaffolding domain,and facilitate cell migration[15,16].
Polymerase 1 and transcript release factor also known as cavin-1 is an essential component of caveolae; it regulates membrane curvature by stabilizing cav-1 in caveolae.The loss of cavin-1 results in the loss of caveolae,and cav-1 release into the plasma membrane.Importantly,cav-1 is required for cavin-1 recruitment to plasma membrane[17,18].In a carotid artery injury model,the local loss of cavin-1 is reported to promote neointima formation.Furthermore,in cultured vascular SMC,the overexpression of cavin-1 suppresses SMC proliferation and migration,whereas its inhibition promotes cell proliferation and migration[19].
The foregoing studies indicate that the loss of cavin-1 and p-cav-1 expression contribute to cell migration,and thus,facilitate neointima formation.Our aim was to elucidate whether similar changes occur in PH that might explain the mechanism of neointima formation.We analyzed the expression of cav-1,p-cav-1 and cav-1-related proteins cavin-1,caveolin-2 (cav-2),vascular endothelial cadherin (VE-Cad),and p-ERK1/2 in rats treated with MCT,hypobaric hypoxia and a group treated with MCT and exposed to hypoxia.
MATERIALS AND METHODS
Ethics statement
Protocols were approved by the Institutional Animal Care and Use Committee of New York Medical College,and conform to the guiding principles for the use and care of laboratory animals of the American Physiological Society and the National Institutes of Health.ARRIVE guidelines were adopted.
Experimental protocol
Male Sprague-Dawley rats (age 6-8 wk,weight 150-175 g,Charles River Wilmington,MA) were allowed to acclimatize in our animal facility for 5 days before the start of the experimental protocols.They were allowed free access to laboratory chow and water.Rats were divided in 4 groups (6-8/group): Group 1; control rats (C)maintained in room air; Group 2,rats received a single subcutaneous injection of MCT(40 mg/kg,M),and kept in room air.Group 3,rats in this group were subjected to hypobaric hypoxia (atmospheric pressure 380 mmHg,H); Group 4,rats in this group received MCT 40 mg/kg and were subjected to hypobaric hypoxia starting on day 1(M+H).We maintained 2-3 similarly treated rats in a cage.The hypoxia chamber was opened twice per week for 15 min to weigh the rats,replenish food and water,and to provide clean bedding.The rats were maintained in our Animal Facility; four weeks later,these rats were brought to the Laboratory for hemodynamic measurements and tissue collection.
Chemicals and antibodies
All chemicals including MCT were purchased from Sigma Aldrich,St Louis,MO.Antibodies: caveolin-1 (cav-1) (sc-894,polyclonal,sc-535641,monoclonal) and VECadherin(VE-Cad) (sc-64580) were bought from Santa Cruz laboratories,CA,United States; Tyr14 Phospho-Cav-1(610684) and Caveolin-2 (610684) were obtained from BD Transduction Laboratories,Palo Alto,CA; Cavin-1 (ab48824) from Abcam laboratories and p-ERK1/2 (4370) and ERK1/2 (4694) from Cell Signaling,Beverley,MA,and βactin (A5441) from Sigma Aldrich.Secondary antibodies,Alexa 488 (donkey antirabbit,green color) and Alexa 594 (donkey anti-mouse,red color) were purchased from Molecular Probes,Eugene,OR.
Measurement of right ventricular systolic pressure
On the day of experiment,rats were anesthetized with an intraperitoneal injection of ketamine (60 mg/kg) and xylazine (5 mg/kg),as approved by our Institution.The trachea was exposed through an incision in the neck,and cannulated with PE 240 tubing,and the rat ventilated in room air at a rate of 70-80 breaths/min.The chest was opened,PE 50 tubing inserted into the right ventricle (RV),and the pressure recorded on a Grass polygraph (model 7E).At the end of the pressure measurements,the lungs were perfused with autoclaved normal saline to remove blood.The left lung and the heart were placed in 10% buffered formalin.The right lung was quickly frozen in liquid nitrogen and stored at -80oC for protein extraction at a later date.
Assessment of RVH
A week later,the heart was removed from formalin and atria trimmed.The free wall of the RV was separated from the left ventricle and the septum (LV).The ratio of RV/LV was calculated to assess RVH.
Protein extraction and western blot analysis
Western blot analysis was carried out as previously described[7].Briefly,the Lung tissue was homogenized in a buffer containing 0.1M PBS (pH 7.4),0.5% sodium deoxycholate,1% igepal,0.1% SDS,and 10 μL/mL phenylmethylsulfonic fluoride,25 μg/mL aprotinin,and 25 μg/mL leupeptin,and phosphatase inhibitor cocktail 1 (10 μL/mL) were added to homogenates placed on ice for 30 min,and centrifuged at 15000 rpm for 20 min at 4oC.Protein concentrations in the supernatants were analyzed by Lowry’s method,using serum albumen as standard (Bio-Rad kit).Fifty or 100 μg of protein from lung supernatants were loaded and separated on a 10%sodium SDS-polyacrylamide gel (Mini Protean-II,Bio-Rad) and transferred to nitrocellulose membrane (hybond ECL,Amersham BioSciences,Piscataway,NJ,United States) using Semi Dry Transfer Cell (Bio-Rad).The membranes were blocked with 5% nonfat milk powder in Tris-Buffered-saline with Tween buffer (TBST,10 mM Tris-HCl,pH 7.4,150 mM NaCl,0.05% Tween 20) at room temperature for one hour.Membranes were then incubated with p-cav-1 (1:500),cav-1 (1:7000),caveolin-2(1:500),cavin-1 (1: 2000),VE-Cad (1:500),or p-Erk1/2 (1:2000),overnight at 4oC.The membranes were washed for 10 min X 3 with TBST and incubated with appropriate secondary antibody for 1 hour at room temperature,then washed for 10 minX3 with TBST buffer.The membranes were reprobed with β actin (1:10000) or Erk (1:2000) as appropriate to assess protein loading.The relative expression of the proteins was quantified using densitometric scanning and is expressed as % normal.
Lung histology and double immunofluorescence
Formalin preserved lung tissues were processed for paraffin blocks.Five to 6 μm sections were cut and stained with H&E stain for the evaluation of pulmonary vessels.Double immunofluorescence was carried out as described previously[7,12].Briefly,lung sections were deparaffinized in xylene (5 min X 2),rehydrated through a range of aqueous ethyl alcohol solution in H2O and immersed in PBS for 5 min.Antigen retrieval was performed by incubating the sections in 10 mM citrate buffer (pH 6),in a microwave oven for 5 min.The slides were then incubated in blocking solution (5%normal donkey serum,0.5% Triton in PBS) for 1 hr at room temperature,followed by an overnight incubation at 4oC with the primary antibody (cav-1α,1:100) in blocking solution.The next day,the slides were washed with PBS for 10 minX3 and incubated in the secondary antibody,Alexa 488 (1:300,green color) at room temperature in a dark place for 1 hr followed by washing in PBS for 30 minX3.For double immunostaining,the sections were blocked again,as described earlier,and incubated overnight in the primary antibody smooth muscle α-actin (1:15); the procedure was repeated using the secondary antibody (Alexa 594,1:200,red color).The sections were examined with a laser scanning confocal fluorescence microscope (MRC 1000,Bio-Rad).
Statistical analysis
The data are expressed as means ± SE.For statistical analysis,one-way analysis of variance followed by Scheffe’s multiple comparison tests was used.Differences were considered statistically significant atP< 0.05.
RESULTS
Weight gain
The four-week weight gain in the control group was 188±11 g.The hypoxia group gained 181 g ± 20 g (not significantly different compared with the controls).The weight gain in the M group was 130 g ± 17 g (P< 0.05vsthe control group).In the M+H group,the weight gain was significantly reduced (45 g ± 17 g,P< 0.05vsControls,H and M groups).Two out of 8 rats in the M + H group lost weight during the 4-wk period.
趾墩和消能墩结合在一起,通过试验研究,证明辅助消能工的消能效果更加明显,这种辅助消能工的消力池就是美国垦务局推荐的USBR-Ⅲ型消力池。
Hemodynamic data and lung histology
As shown in Figure 1,both M and H group revealed significantly increased right ventricular systolic pressure (RVSP) and RVH compared with the controls.RVSP and RVH were significantly higher in the M+H group compared with M and H groups.Significant medial thickness was present in the M and H group.In the M+H groups there was a further increase in medial thickening including the presence of neointimal lesion,and complete occlusion of lumens of smaller arteries (Figure 2).
Localization of cav-1
Double immunofluorescence studies were conducted to visualize the expression of cav-1 and smooth muscle α-actin.The expression of cav-1 is significantly decreased in the larger arteries (95-111 µm) from the M and M+H groups; and enhanced expression of cav-1 in SMC is observed in the M+H group (Figure 3A).Smaller arteries (21-41 µm) from M and M+H groups exhibited a significant loss of endothelial cav-1 accompanied by enhanced expression of cav-1 in SMC (Figure 3B).It is significant,that in the H group,despite increased pulmonary artery pressure,the endothelial cav-1 is not lost,nor is there enhanced expression of cav-1 in SMC.
Expression of cav-1 and p-cav-1
Consistent with our previous observations[12],western blot analysis revealed a significant loss of cav-1 expression in the M group,but it is closer to normal in the M+H group (Figure 4).As shown in Figure 4,the expression of p-cav-1 is increased in the M and M+H groups.The H group,however,did not exhibit a loss of endothelial cav-1 or enhanced cav-1 expression in SMC,or the expression of p-cav-1.
Expression of cav-2 and cavin-1
In the present study,a significant loss of cav-2 is observed in the M and M+H groups(Figure 5A).Similarly,as shown in Figure 5B,there is a significant loss of cavin-1 expression in M and M+H groups.Hypoxia alone has no effect on the expression of cav-2 or cavin-1.
Expression of VE-Cad
Since MCT-induced PH is associated with the progressive loss of endothelial cav-1;we examined the VE-Cad expression in the lungs.The expression of VE-Cad is significantly reduced in the M and M+H groups compared with controls,but it is not altered in the H group (Figure 6).
Activation of pERK1/2
As shown in Figure 7,the expression of p-ERK1/2 is increased in the experimental groups (M,H and M+H).
DISCUSSION

Figure 1 There is a significant increase in the right ventricular systolic pressure (RVSP) in the Monocrotaline (MCT,M) and hypoxia (H) groups compared with the controls (C),and a further increase in the MCT + hypoxia (M+H) group (1A,n=7-8).
The significant results of the present study are that MCT-treated rats exposed to hypoxia (M+H) develop higher RVSP and RVH compared with hypoxia (H) and MCT(M) alone groups; and furthermore,in the M+H group,neointimal lesions and occlusion of small arteries are seen.Significant loss of endothelial cav-1,cav-2,VECad and cavin-1 proteins is observed in the M and M+H groups,indicating extensive EC disruption/damage.In addition,M and M+H groups exhibit enhanced expression of cav-1 in SMC accompanied by p-cav-1 expression.In all experimental groups,expression of p-ERK1/2 is increased.In the H group,there are no alterations in the expression of endothelial cav-1or cav-1-related proteins indicating that there is no EC disruption; nor is there enhanced expression of cav-1 in SMC or the presence of p-cav-1.
Progressive endothelial damage accompanied by endothelial cav-1 loss/dysfunction plays a critical role in the pathogenesis of PH.Cav-1 regulates a number of signal transduction pathways that participate in cell proliferation,cell cycle and apoptosis.Modulation of signaling by cav-1 has an important implication in the development and progression of PH[20].Cav-1 knockout mice develop PH,that is attenuated by cav-1 re-expression[21,22].Cav-1 polymorphism has been shown to be associated with PAH[23].As stated earlier,endothelial cav-1 loss has been observed in idiopathic PAH,heritable PAH and PAH associated with congenital heart defect and drug toxicity[10-12].In the MCT model,we have previously shown the loss of endothelial cav-1 accompanied by a loss of several endothelial membrane proteins such as Tie2,PECAM-1 and soluble guanylate cyclase,indicative of extensive endothelial cell membrane damage[7].PECAM-1 supports EC integrity and maintains barrier function[24].In the present study,significant loss of VE-Cad is observed in the M and M+H groups.VE-Cad belongs to a family of calcium-dependent adhesion proteins responsible for adherence junction and barrier structure.It is tissue specific for EC and is expressed at the intercellular clefts of EC; and mediates cell-cell adhesion,maintains barrier function and contributes to the inhibition of cell growth[25-27].VECad provides EC junctional stability and controls vascular permeability.It interacts with various growth receptors and regulates endothelial proliferative signaling; and mediates contact inhibition of cell growth[28].Furthermore,depletion of cav-1 reduces VE-Cad levels and facilitates endothelial cell permeability[29,30].
Consistent with our previous studies,a significant loss of cav-2 expression is present in M and M+H groups,but not in the H group[12].In all cell types,cav-2 has been shown to be present associated with cav-1.It requires cav-1 for its transport from Golgi body to the plasma membrane.In the absence of cav-1,cav-2 is degraded; and its loss promotes increased cell proliferation[31,32].Cav-2 knockout-mice have been shown to display increased proliferation of EC,hyper-cellular lung parenchyma,and cell cycle progression.Furthermore,cav-2 is thought to suppress ERK1/2 pathway[32,33].Thus,the loss of cav-2 and VE-Cad would have negative effect on membrane permeability,and facilitate cell proliferation; thus,aggravate pulmonary vascular pathology.Interestingly,in hypoxia-induced PH,despite normal endothelial cav-1 expression,an increased activation of p-ERK1/2 is observed,suggestive of cav-1 dysfunction in hypoxia-induced PH.
Caveolae are specialized micro-domains,forming small invaginations.Caveolae act as platforms for multiple signaling activities.Cav-1 is the major protein in caveolae.Cav-1 recruits cavin-1 to the membrane; and cavin-1 is essential for caveolae formation and the stabilization of cav-1[17,18].Loss of cav-1 is accompanied by a marked loss of cav-2 and partial reduction in cavin-1 expression in the lungs.The reexpression of cav-1 rescues cav-2 and cavin-1[34].Cavin-1 knockout mice display lung pathological changes such as remodeled pulmonary vessels and PH.In addition,these mice have been reported to have altered metabolic phenotype,associated with,glucose intolerance,insulin resistance and dyslipidemia[35,36].Furthermore,caveolae act as plasma membrane sensors that respond to stress[37].Caveolae flatten in response to membrane stretch.The flattening is thought to be a protective mechanism; it buffers the membrane and prevents its rupture.However,the flattening of the membrane leads to cav-1 and cavin-1 dissociation[38].Cavin-1 loss has been shown to promote neointima formation; and the over expression of cavin-1 suppresses vascular SMC proliferation and migration[19].In advanced prostate cancer,cav-1 which is present in non-caveolar site because of the lack of cavin-1 is associated with progressive form of the disease; and cavin-1 re-expression neutralizes non-caveolar cav-1 in prostate cancer and reverses its effects,and decreases cell migration[39,40].In the present study,M and M+H models of PH show loss of cavin-1,indicative of loss of caveolae.This would suggest that cav-1 in SMC is in non-caveolar site.Stretchinduced translocation of cav-1 to non-caveolar site in vascular SMC has been shown to mediate ERK activation[41].In addition,it has recently been reported that plasma membrane stretch activates L-type voltage-dependent Ca2+channelsviatyrosine phosphorylated cav-1 and the activation of JNK in mesenteric arterial SMC[42].This observation further supports the view that cav-1 function in non-caveolar site is distinctly different from cav-1 in caveolae[43].

Figure 2 H&E stain of pulmonary arteries from the lungs of control (C),Monocrotaline (MCT,M),hypoxia (H) and MCT+ hypoxia (M+H) groups.
Extensive endothelial cav-1 loss is accompanied by enhanced expression of cav-1 in SMC; and progressive increase in the expression and activity of matrix metalloproteinase 2 at 2 and 4 wk post-MCT[7,44].The present study shows the loss of endothelial cav-1 accompanied by a loss of cav-2,VE-Cad and cavin-1 proteins in the M and M+H groups.These losses are indicative of extensive EC damage and/or loss,which would expose SMC to increased shear stress and strain because of the elevated pulmonary artery pressure.Vascular SMC are reported to be unresponsive to mitogens under normal tensile stress.During altered mechanical stress,protein synthesis in these cells is upregulated in response to growth factors,thus,leading to cell proliferation and neointima formation.Furthermore,during cell cyclic strain,cav-1 is thought to be involved in proliferation signaling,stretch-induced activation and cell cycle entry.Interestingly,exposure of veins to arterial pressure for 15 minutes in wild type mice results in AKT and ERK activation; the response was attenuated in cav-1 knockout mice[45,46].Thus,it is likely that non-caveolar cav-1 in SMC in PH acts as a facilitator of cell migration.
Endothelial cav-1 loss has been shown to result in the activation of proliferative and anti-apoptotic pathways,and as the disease progresses,the continued loss of endothelial cav-1 and EC disruption results in the enhanced expression of cav-1 in SMC,which has been shown to be pro-proliferative[12,13].Importantly,cav-1 contains a tyrosine 14 residue,a target site for phosphorylation by non-receptor tyrosine kinases;and phosphorylation at this site is associated with cell migration[16].Cell migration is essential for neointima formation.Cav-1 has been shown to get phosphorylated at Tyr14 in response to various stimuli.It is thought to be dependent on Src kinase activity and Rho/ROCK signaling.P-cav-1 has been linked to focal adhesion stability and directional cell migration and extracellular matrix remodeling.There is evidence that tyrosine phosphorylated cav-1 is required for cell migration and metastasis; in cancer,upregulated cav-1 correlates with tumor progression.Furthermore,cav-1 phosphorylation has been reported to induce Rho activation and stabilize focal adhesion kinase and promote cell migration[47-49].It is likely that in M and M+H models,enhanced expression of cav-1 in SMC,and increased expression of p-cav-1 coupled with cavin-1 loss may facilitate cell migration and contribute to neointima formation.
In summary,EC integrity and cav-1 play a key role in the maintenance of vascular structure and homeostasis.Our studies underscore the view that endothelial cav-1 dysfunction (in hypoxia model) or endothelial cav-1 loss (in MCT and MCT + hypoxia models) is an important initiating mechanism of PH.In the M and M+H models,progressive disruption of EC and the endothelial cav-1 loss is accompanied by the loss of cav-2,VE-Cad and cavin-1.These alterations are sufficient to activate proliferative pathways such as p-ERK1/2,and facilitate vascular remodeling and PH.Loss of endothelial cav-1 is followed by enhanced expression of cav-1 in SMC and the appearance of p-cav-1.These alterations coupled with the loss of cavin-1 would facilitate cell proliferation,migration and neointima formation,thus progress toward irreversible PH.In the hypoxia-induced PH there was no evidence of EC disruption or loss of endothelial cav-1,cavin-1,cav-2,VE-Cad or enhanced expression of cav-1 in SMC.Despite the presence of endothelial cav-1,p-ERK1/2 is activated indicating that the endothelial cav-1 is dysfunctional.However,EC integrity is maintained during hypoxia-induced PH,which may be the reason why it is reversible on normoxic exposure.
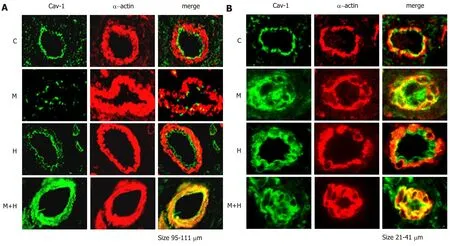
Figure 3 Double immunofluorescence study showing the expression of cav-1 (green) and smooth muscle α-actin (red) in pulmonary arteries from control(C),Monocrotaline (MCT,M),H (hypoxia) and MCT + hypoxia (M+H) groups.

Figure 4 A representative western blot and bar graph depicting the expression of cav-1 and Tyr 14 p-cav-1 in the lungs from controls (C),Monocrotaline(MCT,M),hypoxia (H) and MCT + Hypoxia (M+H) groups (n=5-6).

Figure 5 Representative western blot and bar graph depicting the expression of cav-2 (n=7-8) and cavin1 (n=6-8) in the lungs from controls (C),Monocrotaline (MCT,M),hypoxia (H) and MCT + hypoxia (M+H) groups.

Figure 6 Representative western blot and bar graph depicting the expression of VE-Cad (n=5).

Figure 7 Representative western blot and a bar graph showing the expression of ERK1/2 and p-ERK in controls (C),Monocrotaline (MCT,M),hypoxia (H)and MCT + hypoxia (M+H).
ARTICLE HIGHLIGHTS
Research background
A number of cardiopulmonary and other systemic diseases are known to lead to pulmonary arterial hypertension (PAH),a serious sequel with a high mortality rate.The features of PAH are:impaired vascular relaxation,activation of proliferative pathways; smooth muscle cells (SMC)proliferation,medial hypertrophy,narrowing of the lumen,elevated pulmonary artery pressure and right ventricular hypertrophy (RVH).As the disease progresses,neointima is formed which leads to the irreversibility of PAH.Despite major advances in the field,the cure is not yet available.The diagnosis of PAH is often delayed because of the vague symptoms.This is not surprising because we have previously shown in the monocrotaline (MCT) model of pulmonary hypertension (PH),endothelial damage,progressive loss of endothelial caveolin-1 (a membrane protein) and the activation of proliferative pathways occur before the onset of PH.
Research motivation
By exposing the MCT-treated rats to hypoxia,the disease process is accelerated and by 4 weeks,neointimal lesions are seen,which gives us an excellent opportunity to examine the mechanism of neointima formation.If we can unravel the mechanism of neointima formation,it will allow us to expand the experimental studies to further the knowledge which will aid us in designing curative treatment.
Research objectives
The main objective of the study was to examine the mechanism of neointima formation which renders the diseases irreversible.Our most important observation is that the significant endothelial damage and loss of endothelial caveolin-1 is accompanied by loss of cavin-1 and caveolae,and “enhanced” expression of caveolin-1 in SMC,that is tyrosine phosphorylated.It is possible that these alterations are at least in part responsible for neointima formation.
Research methods
We divided male Sprague-Dawley rats (age 6-8 wks) in 4 groups (6-8/per gr.): Gr.1 Controls,Gr.2 (received a single sc injection of MCT 40 mg/kg),Gr.3 (rats in the group were exposed to hypobaric hypoxia) and Gr.4 (MCT-injected rats were exposed to hypobaric hypoxia starting on day 1).These rats were studied 4 weeks later.Hemodynamic data and RVH were assessed.In addition,we evaluated the histological changes in the pulmonary arteries and the expression of caveolin-1 using immunofluorescence technique.The expressions of caveolin-1,p-caveolin-1,caveolin-2,VE-cadherin (VE-Cad),Cavin-1 and p-Erk1/2 in the lungs were examined using western blot technique.
Research results
Significant loss of caveolin-2,VE-Cad,Cavin-1,indicative of significant endothelial damage was observed in MCT groups.In the MCT group,there was a significant loss of endothelial caveolin-1,and enhanced caveolin-1 expression in SMC in a few arteries; however,the total cav-1 expression in the lungs remained low.In MCT + hypoxia group,a significant loss of endothelial caveolin-1 was accompanied by enhanced expression of caveolin-1 in SMC in a greater number of arteries,which almost normalized the lung caveolin-1 levels.The loss of cavin-1 suggests that the caveolin-1 is not in caveolae,because cavin-1 is essential for the maintenance of caveolar curvature,and in addition,it stabilizes caveolin-1 in caveolae.Furthermore,caveolin-1 is tyrosine phosphorylated in the MCT and MCT + hypoxia groups.ERK activation is observed in all PH models including hypoxia-induced PH.Importantly,there is no loss of caveolin-1,caveolin-2,cavin-1 or VE-Cad in the hypoxia-induce PH.
Research conclusions
Disruption of endothelial cells (EC) and progressive loss of endothelial caveolin-1 results in reciprocal activation of proliferative pathways leading to PH.Extensive damage/loss of EC exposes SMC to increased pressure and shear stress,leading to flattening of caveolae.In addition,caveolin-1 is tyrosine phosphorylated in the MCT and MCT + hypoxia groups.In cancer,loss of cavin-1 and tyrosine phosphorylation of caveolin-1 results in cell migration and metastasis.Thus,the alterations in the caveolin-1 expression in the MCT + hypoxia group may in part facilitate cell proliferation,cell migration and neointima formation.It is important to note,that hypoxia does not alter the expression of caveolin-1 and other membrane proteins; and there is no enhanced expression of caveolin-1 in SMC.However,despite the presence of endothelial caveolin-1,proliferative pathway is activated indicating that the endothelial caveolin-1 is dysfunctional in hypoxia-induced PH.There is no evidence for endothelial membrane disruption in the hypoxia model,which may explain the reported recovery of animals after removal from hypoxia.
Research prospective
The important observation in our study is that the disruption of EC coupled with endothelial caveolin-1 loss initiates pulmonary hypertensive changes and facilitates progression.Further studies are required to determine other factors that might facilitate the “enhanced” caveolin-1 expression in SMC in cell proliferation and migration.We will examine whether cavin-1 and caveolae can be restituted in SMC and normalize caveolin-1 expression,which may abrogate neointima formation.
ACKNOWLEDGEMENTS
This study was in part supported by Cardiovascular Medical Research & Education Fund awarded to RM.
猜你喜欢
中老年保健(2021年12期)2021-11-30
发明与创新(2021年39期)2021-11-05
好日子(2021年8期)2021-11-04
中华诗词(2018年11期)2018-03-26
中学数学杂志(初中版)(2017年4期)2017-08-28
Coco薇(2016年8期)2016-10-09
汽车文摘(2015年11期)2015-12-02
少儿科学周刊·少年版(2015年1期)2015-07-07
中学数学杂志(高中版)(2015年3期)2015-05-28
小说月刊(2014年1期)2014-04-23