
不同水蒸气蒸馏辅助方法对马来沉香挥发油化学组成的影响
2019-05-21 02:47易润青陈兴静杨思惠黄圆圆高晓霞陈晓颖
广东药科大学学报 2019年2期
易润青,陈兴静,杨思惠,黄圆圆,高晓霞,陈晓颖
(1. 广州市医药职业学校,广东 广州 510430; 2.广东药科大学药学院,广东 广州 510006)
沉香既是极其珍贵的药材又是名贵的天然香料,具有浓郁优雅的独特香气[1]。沉香分为进口沉香和国产沉香,进口沉香原植物为瑞香科植物沉香AquilariaagallochaRoxb.[2],主要产于印度、印尼和马来西亚等地;国产沉香原植物为瑞香科植物白木香Aquilaria.sinensis(Lour.) Gigl[3],主要产于我国广东、广西、海南、云南和台湾等省区。
马来沉香AquilariamalaccensisLamk.主产于马来西亚、印尼、泰国,目前,AquilariaagallochaRoxb.已经被归并为AquilariamalaccensisLamk.[4]。马来沉香挥发油主要化学组成为倍半萜类和芳香族类化合物。Jayachandran等[5]采用GC-MS联用技术分析不同等级马来沉香挥发油,结果表明高等级马来沉香油中香橙烯、2-瓦伦烯、白菖烯和斯巴醇等倍半萜类化合物占总气化组分的74.03%,低等级马来沉香中T-杜烯醇、香橙烯、2-瓦伦烯等倍半萜类化合物占总气化成分的43.47%,由此可见,品质好的沉香油倍半萜类化合物相对质量分数更高。
用常规的水蒸气蒸馏方法耗费时间较长,挥发油最终得率小,一般不超过0.1%。为了提高沉香挥发油的得率,王健松等[6]采用CO2超临界流体萃取中山国产沉香木,得油率为1.05%,其中倍半萜类化合物仅占7.39%,2-(2-苯乙基)色酮类化合物占22.678%;邓红梅等[7]采用CO2超临界流体萃取电白的国产沉香油,产率0.62%,其中54.22%为脂肪酸,倍半萜类化合物只占10%左右。梅文莉等[8]用乙醚提取5种国产沉香挥发油,得率在1.7%~5.0%,主要成分包括倍半萜类化合物(11.9%~33.4%)、芳香族化合物(5.0%~20.8%)和脂肪酸类化合物(12.9%~44.2%)。袁观富等[9]采用低温微波提取海南国产沉香,得率约为0.6%,其中约40%为十氢柰型倍半萜类化合物。陈志雄等[10]采用动态微波提取市售沉香木,得率为0.68%。微波提取所得粗提物中含有大量胶质,需用吸附剂除去。李明月等[11]采用果皮酶处理沉香木后再用乙醇索氏提取沉香挥发油,浓缩干燥后称重计算,得率高达21.33%,其中80%以上为2-(2-苯乙基)色酮类化合物。由此可见,除水蒸气蒸馏外的提取方法所得沉香挥发油虽然得率提高了,但其中倍半萜类化合物的含量大多不高,所得沉香挥发油的品质欠佳。
弓宝等[12]采用纤维素酶酶解后石油醚提取国产沉香油,得率从0.062%上升至0.1621%;刘东峰等[13]采用复合酶酶解后的微波提取来萃取沉香油,得率能达到1%左右;唐显[14]采用复合菌种发酵液浸泡沉香木15 d后再用水蒸气蒸馏提取沉香油,平均得率从0.23%提高到0.48%。由此可见,采用酶解、微波、发酵液浸泡等方法可提高沉香油的得率,但这几种辅助方法对沉香挥发油化学组成的影响情况未见报道。
本文以水蒸气蒸馏为基础,分别采用纤维素酶酶解、微波辅助、内生真菌发酵液浸泡、微波结合内生真菌发酵液浸泡不同辅助方法提取马来沉香挥发油,分析各提取方法马来沉香挥发油的化学组成,以期筛选出最佳的马来沉香挥发油提取方法。
1 仪器与试药
1.1 仪器
Agilent 7890A-5975C气相色谱-质谱联用仪;WBFY-205微波化学反应器(予华仪器有限责任公司);HH-6数显电子恒温水浴锅(宏华仪器厂);JA2003A 1/1000电子天平(精天电子仪器)。
1.2 试药
进口沉香1批,经由广东药科大学生物资源与药物开发中心朱爽副教授鉴定为马来沉香AquilariamalaccensisLamk.[15]。按照国家药品监督管理局2004年颁布的《儿茶等43种进口药材质量标准》中的有关规定[2]进行全检,其结果符合进口沉香药材的质量标准。
纤维素酶(上海源叶生物科技公司,酶活性:400 U/mg);BotrysphaeriarhodinaA13发酵液(广东省微生物研究所李浩华副研究员鉴定并提供);正构烷烃混合对照品C7~C40(编号DRH-008SR2,AccuStandard公司,三氯甲烷中1000 mg/L);乙醚(分析纯,广州化学试剂厂);醋酸-醋酸钠缓冲溶液(pH5.0)。
2 方法
2.1 提取方法与供试品溶液的制备
2.1.1 水蒸气蒸馏法(方法一) 精密称取沉香粗粉(过2号筛,且不过3号筛)约20 g,置于500 mL圆底烧瓶内,加入120 mL水,充分振荡摇匀后,静置12 h。将挥发油测定器和冷凝管相连,从冷凝管的上端位置向管中不断加水,使水超过挥发油测定器刻度,当水溢进烧瓶时停止添加。将烧瓶放置在电热套里缓慢加热直到沸腾,同时维持微沸状态8 h左右,静置,冷却。加入少量乙醚收集得到的挥发油,洗涤液置于5 mL量瓶,加乙醚至刻度,摇匀,得供试品溶液1。
2.1.2 纤维素酶酶解辅助水蒸气蒸馏法(方法二) 精密称取沉香粗粉(过2号,且不过3号筛)约20 g和纤维素酶0.024 g,置于500 mL圆底烧瓶内,加醋酸-醋酸钠缓冲液(pH 5.0)100 mL,50 ℃水浴加热,酶解2 h,加水20 mL,自“将挥发油测定器和冷凝管相连”起按“2.1.1”项下方法操作,得供试品溶液2。
2.1.3 微波辅助水蒸气蒸馏法(方法三) 精密称取沉香粗粉(过2号,且不过3号筛)约20 g,置于500 mL圆底烧瓶内,加入120 mL水,微波300 W加热2 min后,静置12 h,自“将挥发油测定器和冷凝管相连”起按“2.1.1”项下方法操作,得供试品溶液3。
2.1.4 内生真菌发酵液浸泡辅助水蒸气蒸馏法(方法四) 精密称取沉香粗粉(过2号,且不过3号筛)约20 g,置于500 mL圆底烧瓶内,加内生真菌发酵液100 mL,35 ℃发酵3 d,加水20 mL,自“将挥发油测定器和冷凝管相连”起按“2.1.1”项下方法操作,得供试品溶液4。
2.1.5 微波结合内生真菌发酵液浸泡辅助水蒸气蒸馏法(方法五) 精密称取沉香粗粉(过2号,且不过3号筛)约20 g,置于500 mL圆底烧瓶内,用少量水湿润后,微波300 W加热2 min,放冷,加内生真菌发酵液100 mL,35 ℃发酵3 d后,加水20 mL,自“将挥发油测定器和冷凝管相连”起按“2.1.1”项下方法操作,得供试品溶液5。
2.2 正构烷烃混合对照品溶液的制备
精密移取正构烷烃C7~C40混合对照品20 μL置2 mL量瓶中,用三氯甲烷定容至刻度,摇匀,制成10 μg/mL的混合对照品溶液,-20 ℃避光保存,备用。
2.3 色谱条件与质谱条件
2.3.1 色谱条件 色谱柱为HP-5 MS毛细管柱(30 m×0.25 mm×0.25 μm);进样口温度:260 ℃;载气:高纯度氦气;流速:1.0 mL/min;进样量:1.0 μL;进样采用不分流方式。升温程序:90 ℃,保温4 min,以1.5 ℃/min升温至130 ℃,保温20 min后以0.5 ℃/min升温至160 ℃,保温5 min后以1 ℃/min升温至180 ℃,保温5 min后以2 ℃/min升温至230 ℃,保温30 min。
2.3.2 质谱条件 调谐方式:标准调谐方式;离子源:EI;离子源温度:230 ℃;电子能量:70 eV;电离电压:1.0 kV;接口温度:240 ℃;质量扫描范围:50~500m/z,溶剂延迟时间:5 min。
2.4 样品测定
依照“2.3”项下的色谱与质谱条件对“2.1”项下的5种供试品溶液进行测定。
2.5 数据处理
将正构烷烃混合对照品(C7~C40)溶液和供试品溶液在同一色谱-质谱条件下进样。通过仪器自带的GC-MS Postrun Analysis进行积分处理,得峰面积。分别求出各类成分以及试样的总峰面积,完成整理对比。用自动质谱退卷积定性系统(Automated Mass Spectral Deconvolution and Identification System,AMDIS)处理样品的总离子流图,去除噪音和图谱重叠的干扰,结合NIST05数据库标准质谱信息比对和保留指数RI(Retention Index)校正,进行组分的鉴定。
3 结果与分析
不同辅助提取方法制备的沉香挥发油供试品GC-MS总离子流图如图1所示,各色谱图总峰面积比较结果见图2。可见,4种辅助水蒸气蒸馏提取方法均能提高挥发油总量,其中纤维素酶酶解辅助提高最多,其次为微波结合内生真菌发酵液浸泡。
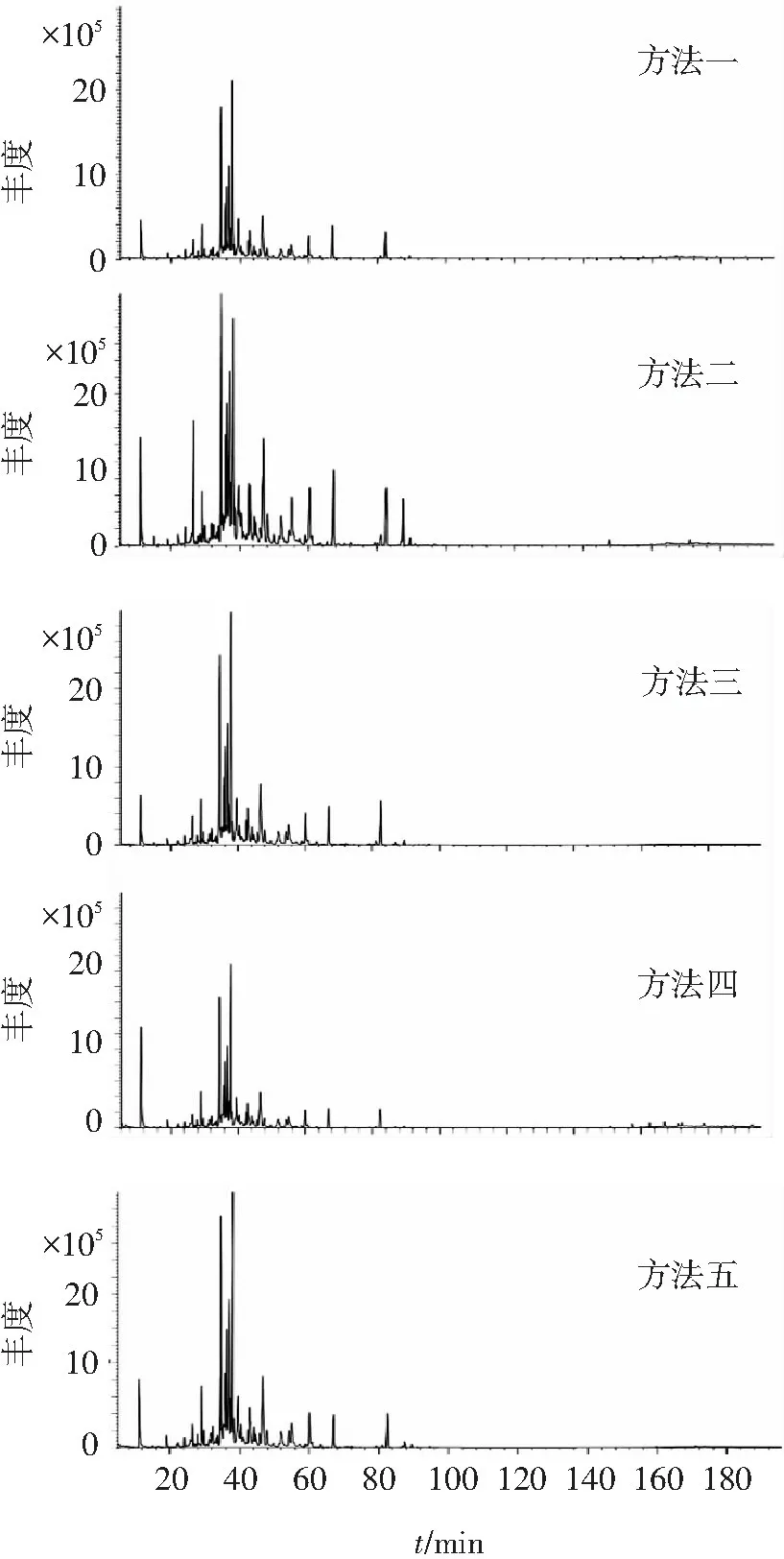
图1不同辅助提取方法的GC-MS总离子流图
Figure1Total GC-MS ion flow diagrams of different auxiliary extraction methods
由表1可见,不同提取方法制备的沉香挥发油的出峰数目分别为105、133、99、111、103;检出物个数均为30;检出物总相对质量分数分别为73.157%、67.381%、74.173%、75.798%、76.524%;其中芳香族类化合物分别为4.258%、4.519%、3.883%、10.425%、4.662%;倍半萜类化合物分别为68.542%、62.702%、69.951%、64.975%、71.334%;倍半萜类化合物总相对质量分数最高的是微波结合内生真菌发酵液浸泡辅助方法(71.334%),最低的是纤维素酶酶解辅助提取方法(62.702%)。
将表1中各方法中相对质量分数最高的7种化合物进行比较,结果见图3。由图3可见,5种方法所得沉香挥发油中质量分数最高的已检成分均为喇叭醇。除内生真菌发酵液浸泡方法中苄基丙酮质量分数较其他方法稍高外,其他4种方法相对质量分数较高的依次为γ-桉叶醇、马兜铃烯、桉烷型、沉香螺醇、苄基丙酮、愈创木烯。由此可见,4种辅助水蒸气蒸馏提取方法所得沉香挥发油的化学成分组成与水蒸气蒸馏方法差别不大。

图2不同辅助提取方法所得GC-MS色谱总峰面积比较图
Figure2The comparison of total peak area of GC-MS chromatogram obtained by different auxiliary extraction methods
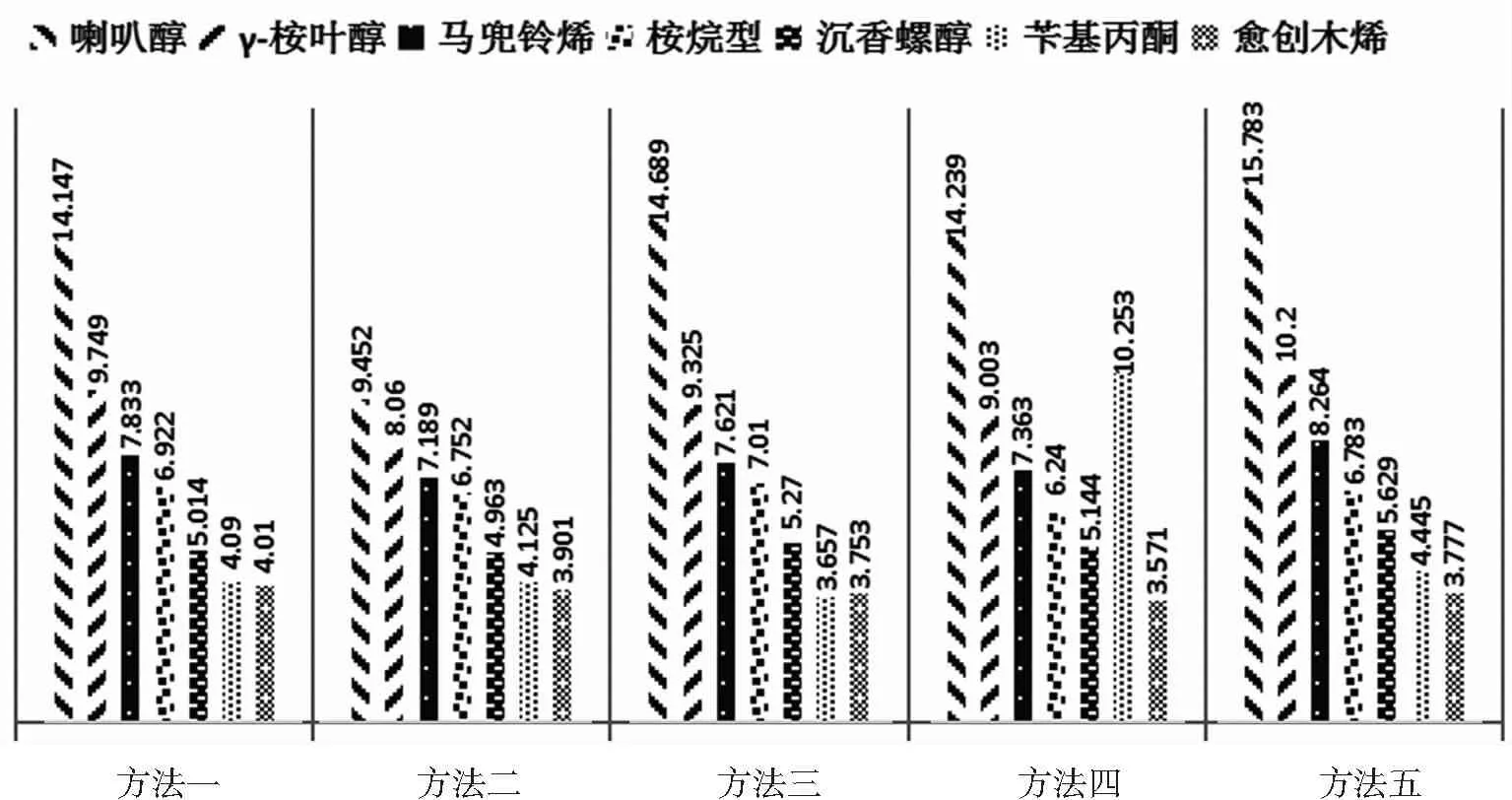
图3不同辅助提取方法中沉香挥发油主要化合物的相对质量分数比较图
Figure3The comparison of the relative percentages of the main compounds of the volatile oil of agarwood by different auxiliary extraction methods

表1 不同辅助提取方法沉香精油样品的成分鉴定及相对质量分数Table 1 Identification and relative content of volatile oil samples from different extraction methods

表1 (续)
注:[a]为NIST库中检索的化合物的RI值;[b]为计算的RI值。
4 讨论
本文5种提取方法获得的沉香挥发油中主要成分均为倍半萜类化合物,其中质量分数较高的γ-桉叶醇、沉香螺醇、α-愈创木烯、苄基丙酮和榄香醇为马来沉香挥发油的主要成分[16],倍半萜类化合物占气化组分的70%左右,接近Jayachandran等[5]划分的高等级马来沉香挥发油(倍半萜类化合物占总气化组分的74.03%)。
纤维素酶酶解辅助水蒸气蒸馏可以通过酶解破坏细胞壁,达到有效增加挥发油提取量的目的,但酶解操作繁琐,提取过程会出现大量的泡沫,增加提取工艺难度。微波是利用微波辐射破坏样品的细胞壁,使挥发油溶出,具有穿透力强、选择性高、加热效率高、受热时间短等特点,可以极大加快反应速度,缩短反应时间,有效提高得率[17-18]。菌群与沉香粉末混合发酵,可大幅度提高沉香油的提取率[14]。本文研究结果表明,微波辅助、内生真菌发酵液浸泡及微波结合内生真菌发酵液浸泡均能增加沉香挥发油的提取量,其中微波加热结合内生真菌发酵液浸泡辅助水蒸气蒸馏法提取的倍半萜相对质量分数最高,在马来沉香挥发油的提取中可发挥较好的作用,但具体提取工艺尚待进一步优化。
致谢:文中所用沉香样品由马来西亚 Feng Yang Biontech(M) Sdn.Bhd吴光明先生提供。
猜你喜欢
分子催化(2022年1期)2022-11-02
世界科学技术-中医药现代化(2021年12期)2021-04-19
中成药(2020年2期)2020-05-12
中成药(2018年12期)2018-12-29
中成药(2017年10期)2017-11-16
小学生学习指导(低年级)(2017年11期)2017-10-23
中学生(2017年13期)2017-06-15
中南大学学报(自然科学版)(2016年2期)2017-01-19
——青蒿素
中国学术期刊文摘(2015年21期)2015-12-24
传奇故事(破茧成蝶)(2015年1期)2015-02-28
