
水溶液中四种脱氧核苷酸与水合铀酰离子相互作用的理论计算
2019-09-09 08:08牟永晓曹建平陈媛媛王朝杰
无机化学学报 2019年9期
牟永晓 曹建平 陈媛媛 卫 涛 王朝杰
(温州医科大学药学院,温州 325035)
0 引 言
随着核能事业的高速发展,人类对能源的需求量与日俱增,其中以铀(U)为基础的核能发电占比越来越大,随之产生的核废料也在不断增加。核废料具有成分复杂、化学毒性和放射性强以及半衰期长达数十万年的特性,因此如何安全转化和处置核废料,同时如何防范核泄漏等问题一直备受关注。但是,目前对铀生物毒性的化学机制认识远不如放射性机制,而DNA是生命体系中储存和传递遗传信息最重要的分子,铀酰离子(UO22+)是铀在水体环境中最稳定的存在形式,且在六价锕酰阳离子中尤为突出,故对铀酰离子与DNA片段相互作用的研究对理解铀酰离子在生物环境中的迁移以及合成对铀酰有较高络合能力的功能核酸分子具有重要的意义。
至今针对铀酰离子对DNA分子结构及正常生理功能影响的研究已广为报道。研究显示,铀在生物体内的化学毒性远大于其放射性毒性,低浓度下铀酰离子可引起生物毒性并造成DNA和多肽结构的氧化损伤[1]。朱寿彭[2]等发现浓缩铀(尤其是可溶性的UO2F2)可诱发精原细胞染色体断片,并随着摄入量的增加,染色体断片发生率随之增高,浓缩铀还可诱发初级精母细胞染色体畸变和精子DNA链断裂。Nielsen课题组[3]在420 nm可见光条件下,发现UO22+能催化单链DNA产生缺口,其机制是UO22+与脱氧核苷酸的磷酸基团结合进而破坏了DNA结构的完整性。除了铀酰离子对DNA分子正常生理功能影响的研究,近年来功能核酸也广泛应用于环境水体中铀元素的检测。李弘杨等[4]报道了基于铀酰离子特异性脱氧核酶的荧光分光光度法检测铀酰离子。Tang[5]等利用二茂铁修饰的脱氧核酶通过电化学检测技术实现了对铀酰离子的检测。相比于实验研究,国内外对铀酰离子与DNA基本结构单元作用体系的理论计算研究仍然较少[6-7],两者具体如何发生作用还待细究。
有关核酸分子结构单元的理论计算研究主要集中在其与主族和过渡金属离子的相互作用[8-9]。Mohamadi[7]等首先对化学合成的[UO2(L)(DMSO)2](L为芳酰席夫碱)进行了表征,并结合分子对接方法对该化合物与鲑鱼精子DNA的相互作用进行了研究,发现该化合物与DNA亲和力强,有部分嵌入到了DNA双链小沟中。Terron[10]等报道了金属银与5′-单磷酸胞苷配合物的X射线固态表征,同时采用M06-2X方法计算了银与胞嘧啶的相互作用性质。李振彰[11]等应用B3LYP方法研究了铬(Cr)金属团簇与碱基相互作用体系,发现铬团簇与碱基环上氮原子及氧原子结合形成的铬-碱基复合物较为稳定。张淑琴[12]在B3LYP/6-311++G**(LANL2DZ)水平详细探讨了Cu2+与胞嘧啶(鸟嘌呤)核苷分子相互作用体系的空间构型及其稳定性,研究表明Cu2+易于与胞苷分子以共价作用形成配位键,当形成四配位键且结构处于同一平面时较为稳定。
本文应用密度泛函理论方法M06-2X[13]结合不同的有效核势(effective core potential,ECP),计算了系列由4种单磷酸脱氧核苷酸(deoxynucleotide monophosphate,dNMP)与铀酰离子在水溶液中形成的配离子结构,探索其红外振动光谱和电子结构性质,并考察4种dNMP与铀酰离子的相互作用特征。
1 计算方法
采用明尼苏达系列泛函中的M06-2X计算水合铀酰离子与脱氧核苷酸分子的相互作用体系,该泛函对计算非共价结合配合物的相互作用能、热化学能和异构化能等性质性能较佳[14-17],已有较多研究使用该泛函计算铀酰离子配合物[18-21]。计算中对铀原子分别采用Stuttgart RLC ECP的“大核势”赝势基组[22]和ECP60MWB-SEG价层基组[23],对其它原子(H、C、N、O 和 P)采用 6-31++G(d,p)基组。其中 RLC ECP 对铀内层电子 1s~5s、2p~5p、3d~5d 和 4f共 78 个电子做冻芯处理,外层14个电子(5f36s26p66d17s2)做价电子处理,ECP60MWB-SEG则对铀内层60个电子做冻芯处理,外层的32个电子(5s25p65d105f36s26p66d17s2)为价层电子。应用Gaussian16[24]程序对水合铀酰脱氧核苷酸配离子体系[UO2(dNMP)(H2O)3]2+进行结构优化和振动频率计算,解析频率结果显示无任何虚频,表明计算所得构型稳定。应用Multiwfn3.6程序[25]对配离子U-dNMP和U-OH2键临界点进行QTAIM[26](quantum theory of atoms in molecules)拓扑分析,并通过原子电荷和分子轨道成分分析探讨了在配位前后各片段的电荷转移以及配离子的轨道组成和能级分布情况,根据Lu等[27]研究结果,使用6-31G(d,p)结合小核势基组进行波函数计算分析。采用极化连续介质模型(polarized continuum model,PCM)[28]模拟水溶液对结构的溶剂效应。
2 结果与讨论
2.1 水合铀酰脱氧核苷酸配离子的结构性质
根据碱基的不同,有脱氧腺苷酸(dAMP)、脱氧胸苷酸(dTMP)、脱氧鸟苷酸(dGMP)和脱氧胞苷酸(dCMP)共4种脱氧核苷酸。自然界游离脱氧核苷酸中,磷酸最常见是位于戊糖的C-5′上,故本文主要以 5′-脱氧核苷酸(5′-dNMP)配位进行分析,其化学结构式如图1所示。鉴于脱氧核苷酸分子柔性较大,空间效应明显,首先用GaussView 6.0[29]程序中GMMX分子构象搜索模块分别筛选出 4种5′-dNMP的最低能量构象,并在M06-2X/6-31++G(d,p)水平对水溶液中4种脱氧核苷酸的优势构象进行优化和振动频率计算,其结构绘于支持信息图S1。从图S1中发现磷酸基团P=O键均与碱基环上和戊糖相连的氮原子邻位的C-H键中氢原子形成了P=O…H氢键,其键长范围在0.218 6~0.227 6 nm,同时磷酸基团羟基与戊糖环氧原子(Os)形成了长约0.194 5~0.198 1 nm 的 O-H…Os键 (除了 5′-dGMP结构)。与其它3种脱氧核苷酸不同的是,5′-dGMP分子磷酸基团上P=O键和羟基分别与鸟嘌呤氨基氢(N11-H键)和N3原子作用形成了距离为0.190 1和0.166 6 nm的氢键。由于5′-dNMP中磷酸基团(H2PO4-)的p Ka值为7.20,在水溶液中几乎不发生电离,主要以分子形态存在,故本文以中性的5′-dNMP进行计算。
在锕酰阳离子赤道平面配位的配体数量一般取决于配位键性质和配体的空间体积,以配体与铀原子作用形成四齿至六齿配位的结构最为常见[30-34]。5′-dNMP结构中磷酸基团、戊糖环羟基和碱基3个部分均可与铀酰离子配位形成多种不对称性配离子,同时5′-dNMP分子的柔性和空间效应较强,因此本文对一分子脱氧核苷酸与三水合铀酰离子配位的结构进行计算讨论。采用M06-2X/6-31++G**(RLC ECP)和 M06-2X/6-31++G**(ECP60MWB-SEG)两 种水平计算配离子结构[UO2(dNMP)(H2O)3]2+,以M06-2X/6-31++G**(ECP60MWB-SEG)水平计算得到的吉布斯自由能由低到高的顺序,依次编号为UdN-j(dN代表 4 种 5′-dNMP,分别记为 dA、dT、dG 和 dC,j=1~5)。共计算得到4种UdA稳定结构,5种UdT结构,4种UdG结构,4种UdC结构,2种水平计算得到的结构和能量数据如图S2和表S1所示。下文的分析讨论中括号内数值为M06-2X/6-31++G**(ECP60MWB-SEG)水平的计算结果。

图1 四种单磷酸脱氧核苷酸(5′-dNMP)的结构示意图Fig.1 Schematic structures of the four kinds of deoxynucleotide monophosphate(5′-dNMP)
脱氧核苷酸与三水合铀酰离子的配位形式主要有4种情况:第一种是磷酸基团P=O键与铀原子形成的单齿配位, 如结构 UdA-1、UdG-1、UdC-1和UdT-3,我们发现在所有配离子中以该配位类型作用形成的结构最为稳定(除了UdT体系)。第二种是碱基环上原子参与的配位,结构UdA-2是氮原子与铀原子作用形成的单齿配位,其中2个水分子分别与磷酸基团的羟基和碱基环上的氨基发生了弱相互作用。UdT-1、UdT-2、UdG-2和UdC-2结构是羰基氧与铀原子作用形成的单齿配位,发现羰基氧与铀原子的作用距离比U-OH2配位键稍短。第三种是戊糖环C-3′位羟基氧(O3′)与铀酰离子作用形成的单齿配位,如结构 UdA-3、UdT-4、UdG-3 和 UdC-3,羟基氧与铀原子的作用距离比水分子与铀原子的作用距离稍长。第四种是磷酸基团上2个羟基氧均与铀酰离子作用形成的双齿配位,如结构UdA-4、UdT-5、UdG-4和UdC-4,其配位键距离同样比U-OH2配位键长。与其它3种体系不同的是,UdT体系最稳定构型是以碱基环上的O4原子与铀酰离子配位(UdT-1),而磷酸基团P=O键作用结构(UdT-3)的吉布斯自由能比UdT-1结构高15.4(9.9)kJ·mol-1。2种计算水平得到的构型相对吉布斯自由能顺序大致相同。为比较4种脱氧核苷酸与铀酰离子配位行为的差异,下文对5′-dNMP中磷酸基团P=O键与中心铀原子配位的配离子结构进行详细讨论。
优化得到的四水合铀酰离子及UdN体系在水溶液中的结构如图2所示(以M06-2X/6-31++G**(ECP60MWB-SEG)水平下优化构型为例),几个关键的键长和键角参数列于表1中。计算得到四水合铀酰离子U=O键和U-OH2配位键的平均键长分别为0.169 8(0.172 6)nm和0.251 6(0.243 2)nm,结构中水分子排列类似于“梯形”,与孙晓玲[35]采用M06-2X/6-311++G**(RLC ECP)水平计算得到的水溶液中UO2(H2O)42+构型一致,其U=O键和U-OH2配位键长分别为0.169 4和0.255 7 nm,与本文计算值相接近。 Cao[36]等采用 B3LYP/RLC ECP和 MP2/RLC ECP计算水平得到水溶液中四水合铀酰离子U=O键长分别为0.174 0和0.177 4 nm,U-OH2键键长分别为0.239 7和0.238 2 nm,与本文在大核势下的计算结果相差较大,可能是由于密度泛函方法选取不同。在ECP60MWB-SEG基组水平下的计算结果与通过X光吸收精细结构谱(EXAFS)实验方法[37]测得的U=O键0.176 0 nm、U-OH2键0.241 0 nm符合较好,表明应用小核势基组计算铀酰离子脱氧核苷酸体系是较为合适的。

图2 在M06-2X/6-31++G**(ECP60MWB-SEG)水平水合铀酰脱氧核苷酸配离子在水溶液中的结构Fig.2 Structures of hydrated deoxynucleotides coordination uranyl ions in aqueous phase obtained from the M06-2X/6-31++G**(ECP60MWB-SEG)level

表1 水合铀酰脱氧核苷酸配离子在两种水平上的结构参数,括号中为M06-2X/6-31++G**(ECP60MWB-SEG)方法的计算结果Table 1 Equilibrium structure parameters of hydrated deoxynucleotides coordination uranyl ions at the M06-2X/6-31++G**(RLC ECP)and M06-2X/6-31++G**(ECP60MWB-SEG)levels,the results of the M06-2X/6-31++G**(ECP60MWB-SEG)are in parentheses
对采用ECP60MWB-SEG赝势基组的计算结果进行比较,我们发现UdN体系中一水分子均倾向于与脱氧核苷酸配体发生协同作用。4种配离子的U=O键长值较接近,最大相差不超过0.000 7 nm,平均键长为0.173 0 nm,与UO2(H2O)42+的U=O键长相比有伸长趋势。而U-OdNMP键键长有所不同,最大差值为0.003 9 nm,配离子U-OdNMP键长按照UdA-1(0.237 9 nm)>UdG-1(0.237 8 nm)>UdC-1(0.237 0 nm)>UdT-3(0.234 0 nm)的顺序递减。 文献[38]采用B3LYP/6-31++G*(RSC ECP)方法计算水溶液中三水合铀酰氨甲酰磷酰胺酸配合物体系[UO2(CPO)(H2O)3]2+,配合物结构中U-OP键长0.238 0 nm,与本文在小核势基组水平得到的配离子结构U-OdNMP键键长相接近。优化得到U-OH2键长在0.246 9~0.253 5 nm范围内,这一距离在已报道的水合铀酰配合物UOH2距离范围内[39-42],计算结果显示UdT-3配离子的U-OH2键最长,UdC-1配离子的U-OH2键最短。4种配离子的O=U=O角度在171.3°~177.0°之间,近似为线形结构,以UdA-1配离子的O=U=O键角最小。对两种基组水平的计算结果进行比较,ECP60MWB-SEG基组计算U=O键长比RLC ECP基组计算值长约0.002 8 nm,但配位键键长比RLC ECP基组计算值短,表明在ECP60MWB-SEG基组下配位键合更强,与辜家芳等[43]分别通过大核和小核有效赝势基组计算环辛四烯与金属铀有机配合物的结果相一致。
2.2 水合铀酰脱氧核苷酸配离子的能量分析
为了分析比较4种配离子稳定性及脱氧核苷酸与水合铀酰离子的结合强度,并考察形成配离子前后脱氧核苷酸由于形变所产生的能量变化。我们分别计算了UdN体系的总结合能(ETotal),脱氧核苷酸的变形能(ED),计算公式如下:


表2 水合铀酰脱氧核苷酸配离子在两种水平上的结合能和变形能Table 2 Binding energies and deforming energies of hydrated deoxynucleotides coordination uranyl ions at the M06-2X/6-31++G**(RLC ECP)and M06-2X/6-31++G**(ECP60MWB-SEG)levels
除了总结合能,UdT-3结构的单结合能也较其它3种配离子的低,这是由于UdT-3结构中的一分子水与磷酸的羟基氧作用形成氢键,而另3种配离子中一分子水是与碱基环上氮原子和(或)戊糖环氧原子作用形成了氢键,前者形成的氢键强度相对较弱。对比2种赝势基组的计算结果,ECP60MWBSEG基组计算的配离子单结合能数值均比RLC ECP基组的计算值大,与前文U-dNMP配位键键长计算结果相对应,同时2种计算水平得到的单结合能强弱顺序是一致的,按照UdC-1>UdG-1>UdA-1>UdT-3的顺序依次减弱。变形能数值最小是5′-dAMP,说明5′-dAMP在形成配离子后构型变化最小。比较不同配位类型的总结合能(表S1),结果表明,磷酸基团P=O参与配位时,配离子总结合能最大,而当磷酸基团上双羟基氧进行配位时,总结合能最小。
2.3 水合铀酰脱氧核苷酸配离子的振动光谱
UdN体系的红外光谱模拟图如图3所示,图中标注了 U=O 键对称(νss)和反对称(νas)伸缩振动频率及P=O键的伸缩振动频率。例如在ECP60MWBSEG赝势基组下,UdA-1结构970和1 021 cm-1处吸收峰分别归属于U=O键的对称和反对称伸缩振动,1 223 cm-1处吸收峰归属于P=O键的伸缩振动。U=O键反对称伸缩振动峰强度明显高于对称伸缩振动峰强度,与文献[36]报道的结果一致。计算得到在ECP60MWB-SEG基组下四水合铀酰离子U=O键对称和反对称振动频率分别为1 004和1 050 cm-1,文献[48-49]应用EXAFS方法测定五水合铀酰离子U=O键对称和反对称振动频率分别为869和965 cm-1,比本文计算值分别低了135和85 cm-1,其原因是理论方法的不同会对频率计算造成系统性偏差,同时随着铀酰赤道面配体数的增加,U=O键伸缩振动频率逐渐减弱[6]。

图3 水合铀酰脱氧核苷酸配离子在两种水平上的红外光谱图Fig.3 Infrared spectra of hydrated deoxynucleotides coordination uranyl ions at the M06-2X/6-31++G**(RLC ECP)and M06-2X/6-31++G**(ECP60MWB-SEG)levels
与四水合铀酰离子U=O键的伸缩振动频率相比,UdN体系中U=O键伸缩振动频率均发生红移,以UdA-1配离子变化程度最大,对称和反对称伸缩振动频率分别红移了34和29 cm-1。4种配离子U=O键的对称伸缩振动频率按照UdA-1(970 cm-1)<UdC-1(980 cm-1)<UdG-1(982 cm-1)<UdT-3(991 cm-1)的顺序递增,表明对应U=O键变强,键长逐渐减小,与上文给出的键长结果相对应。文献[50]表明U=O键的振动频率红移程度越大,则配合物稳定性越强,结合前文结合能分析,我们得到的结果与文献结论一致。与自由状态下5′-dNMP的振动频率值(表S2)相比,形成配离子后P=O键的振动频率均发生红移,红移范围在20~61 cm-1间,表明P=O键强度减弱,其中UdA-1配离子的P=O键红移程度最大,红移最小的是UdC-1配离子。2种计算水平的结果表明,RLC ECP基组下计算得到的红外振动频率值稍大于ECP60MWB-SEG赝势基组下的计算结果。
2.4 电子密度拓扑分析
为进一步分析配离子中U-OdNMP和U-OH2配位键性质和强度,我们对其进行QTAIM分析。根据QTAIM理论[26],2个相互作用原子之间会有一键临界点(BCP),它是电子密度曲线上的鞍点,对应于电子对电子密度的最小值。当BCP处的电子密度ρ(r)大于0.2且拉普拉斯电子密度▽2ρ(r)小于0时,说明该配位键具有共价键性质(开壳层作用),反之当ρ(r)小于0.2且▽2ρ(r)大于0时,该配位键具有离子键性质(闭壳层作用)。表3为采用2种计算水平得到的U-OdNMP和U-OH2配位键在键临界点处的电子密度和电子密度拉普拉斯值。以ECP60MWB-SEG赝势基组计算结果为例,UdN体系中U-OdNMP键在BCP处的 ρ(r)和▽2ρ(r)分别在 0.061~0.066 和 0.258~0.288范围内,U-OH2键在 BCP 处的 ρ(r)和▽2ρ(r)分别在0.045~0.053 和 0.177~0.208 范围内,说明 U-OdNMP和U-OH2配位键具有离子键性质。U-OdNMP键临界点参数值均比U-OH2键的参数值大,表明5′-dNMP与铀原子的作用键强度大于水分子与铀原子的作用键强度。与四水合铀酰离子U-OH2键的临界点参数值相比,形成配离子后U-OH2键各参数值均减小,表明U-OH2配位键有减弱趋势。2种水平的计算结果的定量差异也较为明显,在RLC ECP基组下计算的临界点参数值偏小。
2.5 电荷转移分析
为考察形成配离子后各片段的电荷转移变化情况,我们将UdN体系分为3个片段(铀酰离子、脱氧核苷酸和水分子)进行Mulliken电荷分析。在2种计算水平下铀酰离子和配体所带的Mulliken电荷于表4中列出。
自由状态下铀酰离子所带电荷为2.000,具较强的吸电子能力,可吸引配体向其转移电子。从表4中可以看出,在配位前后,铀酰离子电荷减少的同时配体片段电荷有所增加。文献[36]采用B3LYP/RLC ECP和MP2/RLC ECP水平计算得到水溶液中四水合铀酰离子 UO22+的Mulliken电荷分别为 1.150和1.440,本文对应的计算值在上述2种水平范围内。比较ECP60MWB-SEG赝势基组计算结果,显示UdA-1中铀酰离子片段带有最少的正电荷,UdC-1配离子次之,表明5′-dAMP和水分子向中心铀酰离子转移电子数最多,共为0.822,而铀酰离子所带电荷数最多的是UdT-3配离子,表明5′-dTMP和水分子向铀酰离子片段转移电子数最少,共为0.767,与前文配离子的总结合能结果相一致,说明配体与铀酰离子之间转移电子数越多,配离子结合强度越大。5′-dNMP失去的电子数比水分子片段失去的电子数多(0.138~0.235),表明 5′-dNMP 更容易转移电子,形成的U-OdNMP配位键亦较强。比较2种水平的计算结果,RLC ECP基组下计算得到的配体间电子转移数较多。
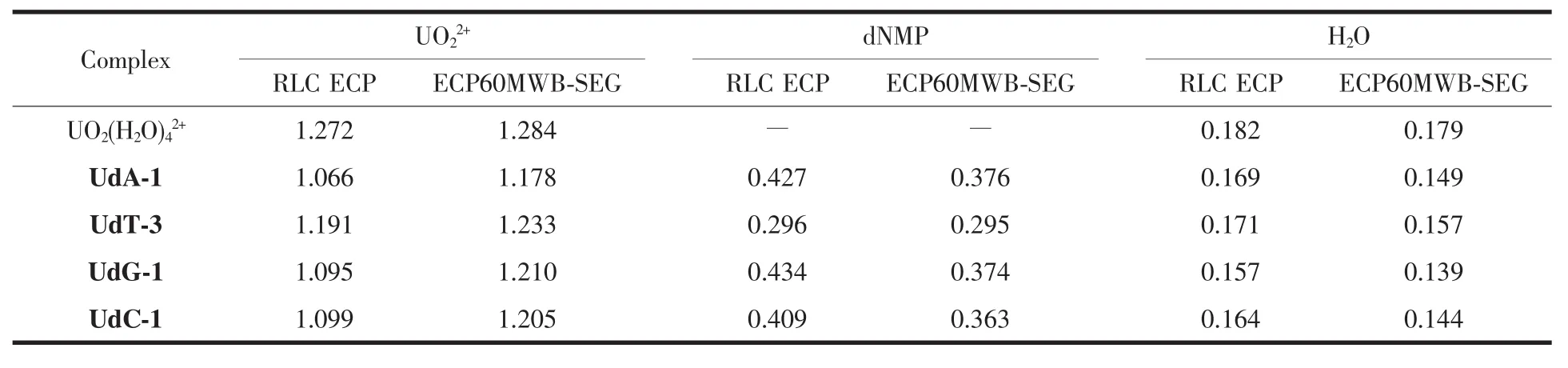
表4 水合铀酰脱氧核苷酸配离子中各配体在两种水平上的Mulliken电荷Table 4 Mulliken charges of each ligand in hydrated deoxynucleotides coordination uranyl ions at the M06-2X/6-31G**(RLC ECP)and M06-2X/6-31G**(ECP60MWB-SEG)levels
2.6 前线分子轨道分析
图4为UdN体系的前线分子轨道能级图,图中Δε表示最高占据轨道和最低空轨道的能级差,表5为使用Mulliken方法计算得到体系原子对前线分子轨道的贡献结果。下文中将最高占据轨道HOMO简写为H,最低空轨道LUMO简写为L。结合图4和表5可知,4种配离子的高占据轨道H~H-1主要为碱基或戊糖环贡献。对于H-1轨道,UdA-1中是碱基环上N1、N3和C5原子各自的2p轨道形成的离域π键,其中N1、N3和C5原子2p壳层的贡献分别为28.6%、14.6%和12.3%。UdC-1配离子H-1轨道与UdA-1类似,同为碱基环上原子的贡献。UdT-3和UdG-1配离子中戊糖环氧原子(Os)和C-3′位羟基氧(O3′)原子2p壳层对H-1轨道有主要贡献,后者鸟嘌呤环上N9原子的2p壳层对此分子轨道也有少量贡献。再如4种配离子的H轨道,主要对应碱基环上双键碳原子2p壳层形成的π键,同时N或O原子的2p壳层也有不同程度的贡献。4种配离子的L~L+5分子轨道基本属于U(5f)轨道(图S3),且配离子的轨道图形类似 (除了UdT-3结构),L~L+1和 L+2~L+3 分别对应为 U(5f啄)和 U(5fφ)轨道,L+4~L+5轨道描述的是U(5fπ)与轴向氧原子的2p轨道作用成键, 而UdT-3结构中 L+1~L+3为 U(5fφ)轨道。有研究显示[51-52],锕系酰基离子(AnO22+)键合作用中5f轨道成分愈多,锕酰离子轴则愈趋近于线形,而本文计算的UdA-1配离子O=U=O键角变化最大,可能与该结构中U(5f)轨道总贡献量最少有关。UdG-1配离子HOMO-LUMO轨道能级差最小,为5.51 eV,UdC-1轨道能级差最大。

图4 在M06-2X/6-31G**(ECP60MWB-SEG)水平上水合铀酰脱氧核苷酸配离子的部分前线分子轨道能级图,轨道等值面为0.04Fig.4 Partial frontier molecular orbitals energy level diagrams of hydrated deoxynucleotides coordination uranyl ions at the M06-2X/6-31G**(ECP60MWB-SEG)level,and the orbital isovalue is set to 0.04

表5 在M06-2X/6-31G**(ECP60MWB-SEG)计算水平水合铀酰脱氧核苷酸配离子中前线分子轨道的组成Table 5 Contribution of hydrated deoxynucleotides coordination uranyl ions to frontier molecular orbitals at the M06-2X/6-31G**(ECP60MWB-SEG)level
3 结 论
运用密度泛函理论方法M06-2X在2种 相对论有效势和 6-31++G(d,p)基组水平研究了[UO2dNMP(H2O)3]2+配合物。对几何结构、振动光谱、结合能、电子密度拓扑参数、原子电荷转移及轨道成分等方面进行了阐述,得到以下几点结论:(1)应用小核势基组可以更好地描述铀酰离子脱氧核苷酸体系的结构特征。(2)脱氧核苷酸与三水合铀酰离子存在4种稳定配位方式,以磷酸基团P=O键与中心铀原子形成的单齿配位结构最稳定 (除了UdT体系)。(3)4种配离子U=O键长值相接近,但配位键距离有所不同。(4)能量分析表明,配离子的总结合能呈UdA-1>UdC-1>UdG-1>UdT-3 的趋势,5′-dAMP 的变形能最小,ECP60MWB-SEG基组计算的结合能均比RLC ECP基组计算值大。(5)红外振动光谱分析发现,配离子中U=O键和P=O键伸缩振动频率均发生红移,且以UdA-1配离子变化程度最大。(6)QTAIM分析表明U-dNMP配位键具有离子键性质,脱氧核苷酸与铀原子的作用键强度大于水分子与铀原子的作用键强度。(7)Mulliken电荷分析发现配体片段向铀酰离子转移了电子,且配体与铀酰离子之间转移电子数越多,形成的配离子结合强度越大。(8)分子轨道分析表明,高占据轨道贡献主要来自脱氧核苷酸配体的π轨道,而低阶空轨道基本为U的5f电子轨道,UdG-1配离子的HOMO-LUMO能级差最小。
铀酰离子与脱氧核苷酸配合物的DFT计算研究结果将为我们进一步拓展其它酰离子与更复杂的生物大分子如核酸片段或多肽结构的作用体系奠定基础。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
腐植酸(2020年5期)2020-12-20
人工晶体学报(2019年5期)2019-06-18
当代陕西(2019年6期)2019-04-17
World Journal of Clinical Cases(2019年4期)2019-04-16
科技创新与品牌(2019年12期)2019-02-06
分析化学(2018年2期)2018-03-02
外语学刊(2014年3期)2014-12-03
