
Double-layered osmotic pump controlled release tablets of actarit:In vitro and in vivo evaluation
2019-09-11 01:19YuennLiHoPnHonglingDunJintingChenZhihongZhuJingxinFnPingfeiLiXinggngYngWeisnPn
关键词:参考文献
Yuenn Li,Ho Pn,Hongling Dun,Jinting Chen,Zhihong Zhu,Jingxin Fn,Pingfei Li,Xinggng Yng,Weisn Pn,*
aShenyang Pharmaceutical University,No.103,Wenhua Road,Shenyang City,Liaoning Province 110016,China
b Liao Ning University,No.66,Chongshan Middle Road,Huanggu District,Shenyang City,Liaoning Province 110036,China
Keywords:Actarit Double-layered Osmotic pump tablet Pharmacokinetics In vivo-in vitro correlation
ABSTRACT The aim of the study was to develop actarit double-layered osmotic pump tablets to overcome the weak points of actarit common tablets,such as short half-life and large plasma concentration fluctuations. Single factor experiment and orthogonal test were applied to optimize the formulation;the pharmacokinetic study was performed in beagle dogs adopting actarit common tablets as reference tablets. The optimal formulation was as follows:drug layer: 150 mg actarit, 240 mg PEO-N80, 50 mg NaCl; push layer: 140 mg PEO-WSR303,20 mg NaCl;coating solution:30 g cellulose acetate and 6 g PEG 4000 in 1000 ml 94%acetone solution,60 mg coating weight gain.The pharmacokinetic study showed that Tmax was prolonged by the contrast of commercial common tablets with constant drug release rate,but the bioavailability was equivalent. And a good in vivo-in vitro correlation of the actarit osmotic pump tablets was also established.The designed actarit osmotic pump tablets can be applied for rheumatoid arthritis,proposing a promising replacement for the marked common products.©2018 Shenyang Pharmaceutical University.Published by Elsevier B.V.This is an open access article under the CC BY-NC-ND license.(http://creativecommons.org/licenses/by-nc-nd/4.0/)
1. Introduction
Rheumatoid arthritis (RA) is an autoimmune disease involving joints,which is prevalent now and can be caused by many reasons but not an exact one [1,2]. RA leads to synovial joint destruction and systemic manifestations but its pathophysiology remains unclear [3,4].The increase of interleukin-1 (IL-1) secretion which is from the single-core-M system of RA patients, is the most important reason of all. Excessive IL-l causes synovial cell proliferation,synovial hypertrophy,stimulation of chondrocytes and the production of prostaglandin E2 collagenase,then will lead to bone destruction.In addition,a large number of auto-antibodies produced by B lymphocyte is also extremely unfavorable to patients. Optimal management of rheumatoid arthritis requires an understanding of the therapeutic goals,and drugs are the one part of it[5].Current therapeutic regimen for RA patients includes non-steroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, diseasemodifying anti-rheumatic drugs(DMARDs)[6,7]and Biological drugs[8].
Actarit[9,10]is a white or almost white crystalline or crystalline powder,slightly soluble in water.Actarit is the diseasemodifying anti-rheumatic drugs and the mechanism of actarit is different from the commonly used anti-rheumatoid drugs which correct the immune system disorders through the immune regulation.Actarit has no significant effect on the production of arthritis and humoral immunity,but can react by immunomodulation of arthritis and regulate cellular immune function.The oral administration of actarit is well tolerated and has a wide safety range.Nevertheless,the common tablets of actarit found in the market, have the deficiencies of short plasma half-life, only 1 h. The common preparation has bad compliance,and clinical researches showed that the common tablets have the plasma concentration fluctuations.It is therefore desired to develop a suitable dosage form to prolong the release of the tablet and avoid the above disadvantages [8,10]. Nowadays, some progress has been made in the research of actarit preparations,such as the actarit injectable solid lipid nanoparticles. It may provide long residence time of the drug and reduce the toxicity.However,it is non-oral administration preparation and has many limitations[11].There are few research about the oral dosage of actarit,only the common tablets were sold in the market but were not popular because the deficiencies of actarit above.
Osmotic pump is an oral drug delivery system that utilizes osmosis pressure to drive drugs out [12,13]. By contrast with the conventional pellets,osmotic pump tablets can maintain the stable plasma concentration and are independent of presence or absence of food,pH of gastrointestinal[14,15].Besides,the drug release of osmotic system is typical zero-order [16].Unlike water-soluble drugs, the water-insoluble drug itself is difficult to dissolve for producing osmotic pressure, thus it must be released by other forces. The core of push-pull osmotic pump tablets is composed of a drug layer containing an active drug and a push layer having expanding agents[17].The core above finally coated with a layer of semi-permeable film and drilled a release orifice at the drug layer. When the moisture in the biological liquid passes through the coating film into the tablet core,the drug-containing layer is hydrated and forms a suspension of the drug. At the same time, the push layer is hydrated and swollen to help the drug release at the state of suspension [18]. Ultimately, the preparation will achieve the release effect we expect.Although there were some researches about the innovative dosage of actarit,such as nanoparticles[11],stability and feasibility of the actual industrial production were difficult.In one word,actarit osmotic pump tablets can not only improve the weakness of the drug itself, but also be easier to enlarge production and quality controllable.Most importantly,patient compliance can be improved.In a word,it has great scientific and commercial value in the future.
Above of all, prepared the actarit double-layered osmotic pump tablets through prescription investigation.Then,in vitro dissolution release and in vivo pharmacokinetics were performed to verify the rationality and reliability of the preparation.
2. Materials and methods
2.1. Materials
Actarit was purchased from Jiangsu Suzhong Pharmaceutical Co.,Ltd (China).Polyoxyethylene was obtained from Dow Chemical Co. (New Jersey, USA). Sodium chloride was purchased from Shenyang Reagent Factory (China). Fine powder Silicone was purchased from Henan Chemical Co., Ltd.(Henan, China). Polyethylene glycol was purchased from Shanghai Chemical Reagent Procurement Supply Station(China).Cellulose acetate was purchased from Shanghai Cellulose acetate plant (China).The remaining reagents were of analytical grade.
2.2. Preparation of the doubled osmotic pump tablets
Drug-layer granules: Mixed the 150 mg 80 mesh screened actarit,low weight polyoxyethylene and sodium chloride evenly.After that,adopted the same amount of incremental method to add the appropriate amount of fine powder silica gel,then mixed well for use.
Push-layer granules:Blended the high weight polyoxyethylene and sodium chloride which were filtered through 80 mesh screen well,then added the powder silica gel using the same method above.
Tableting: Put the appropriate amount of drug-layer granules into the die of the tableting machine, and then pressed it by appropriate pressure.Then filled the push-layer granules above and pressed again.Finally,the entire core of the tablet was finished.
Coating:The tablet core was coated with acetone solution(94%) of 30 g cellulose acetate. The coated product was dried at 40°C for 16 h and drilled a corresponding size orifice at the drug-layer.
Coating solution: 1000 ml coating solution (acetone: water=94:6) was prepared once a time. 30 g cellulose was dissolved in the corresponding volume of acetone at first.At the same time,the polyethylene glycol was dissolved in the prescription volume of water.Finally,the polyethylene glycol solution was slowly poured into the cellulose acetone solution with stirring.
Coating process parameters were as follows: the weight and temperature of coating bed were 100 g and 35°C,respectively. The diameter of the coating pan was 200 mm. Panrotating rate was 40 rpm.The vertical inclination was 30°.The import rate of coating solution was 7 ml/min. The pressure was 6-8 atm.
2.3. Solubility measurement
To obtain the solubility of actarit in various solutions,overdose actarit was added to 0.1 mol/l HCl,pH 6.8 buffer,pH 7.4 buffer and water, respectively. Then the solutions were agitated at 100 rpm in the water bath at 37°C.Except 0.1 mol/l HCl which was demanded to sample after 24 h,the others were sampled every 8 h until the measured value was constant.After filtration and appropriate dilution,the absorbance of samples was measured at 244 nm wavelength by UV spectrophotometer.
2.4. In vitro drug release
The in vitro release study was carried out with paddle method at 37°C. The agitation rate was 50 rpm and nine hundred milliliters of distilled water was used as the release medium for tablets.At the predetermined time intervals,5 ml of samples were withdrawn at 2,4,6,8,10 and 12 h,and replaced by fresh medium of equal volume and same temperature. The samples were then filtered through a 0.8 μm membrane filter and measured appropriate volume of filtrate precisely for diluting. After diluting, the samples were determined to measure the absorbance at 244 nm using an UV spectrophotometer.
The similarity of release profiles was evaluated by similarity factor (f2) recommended by FDA. The f2value was calculated by the following equation[19,20]:
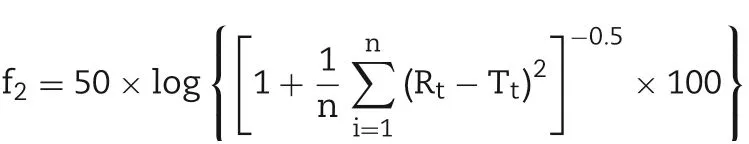
Where Rtand Ttare the accumulated release rates of the reference preparation and test preparation at every time points and n is the number of the time points.It is reasonable to think the two curves were similar when the f2value was higher than 50.
2.5. In vivo pharmacokinetic study in beagle dogs
The protocol of the animal experimental study was approved by Liaoning Medical University Laboratory Animal Ethics Committee.The animals were six healthy beagle dogs weighting 15-18 kg.A two-period crossover experimental design was adopted with the washout period of 10 d and the test beagle dogs were randomly divided into two groups which were fasted for 12 h before administration of reference preparation R(actarit common tablets,100 mg)and test preparation T(actarit self-made tablets, 150 mg). The dogs cannot take other medicine for two weeks before the experiment started.Before the experiment, it was essential to place a retention needle in the foreleg small vein of Beagle dogs,blood samples (5 ml)were withdrawn at different time points after administration.Sampling times of reference tablets were 0,0.5,1,1.5,2,2.5,3,4,6,8,12 h,while the test tablets group was conducted at 0,0.5,1,1.5,2,2.5,3,4,6,8,12,16,20,24 h.Once the blood samples were obtained, they were transferred into heparinized centrifuge tubes immediately. Then centrifuged at 4000 rpm for 10 min and stored at-20°C for analysis.
Chromatographic conditions were as follows: Diamonsil C18 column (4.6 mm×200 mm,5 μm,DIKMA,Beijing,China);mobile phase:acetonitrile-methanol-distilled water(12:16:72,v/v/v) with 0.5 ml trifluoroacetic acid per 1000 ml; flow rate:1.0 ml/min; injection volume: 20 μl; UV detector wavelength:244 nm;room temperature.
Acetaminophen as the internal standard was added into the plasma samples.It was followed by the adding of 0.1 mol/l HCl and dichloromethane, and the vortexing at 4000 rpm for 10 min. The water layer was separated and added ethyl acetate. Then vortexed for mixing and the supernatant was dried with nitrogen.

Table 1-Solubility of actarit in different solvents.
The residue was dissolved by ultrasound with mobile phase and moved into the 1 ml tip centrifuge tube at 10,000 rpm, sustained 5 min. Then injected 20 μl of supernatant into HPMC directly and recorded the peak area. The blood concentrations of beagle dogs were calculated according to standard curve equation.The peak area ratio of actarit to internal standard was linearly regressed by concentration of actarit.Finally,the standard curve equation was y=1.7335C- 0.2569 (r=0.9974) at the range of 0.10-8.08 μg/ml.The LOQ was 0.10 μg/ml.The extraction recovery was 72.5%-75.2%.The relative standard deviations were all less than 10.0%for intraday and inter-day tests.Afterwards,with the help of software DAS2.0,imported the related data such as plasma concentration and the software calculated the pharmacokinetic parameters of reference and test group.Besides,the relative bioavailability and bioequivalence were also obtained.
3. Results and discussions
3.1. Solubility measurement
The solubility of actarit in the various solutions was displayed in Table 1. Actarit is slightly soluble in the four solutions.900 ml of water can meet the leakage conditions. Besides, it was cheap and easy to get.So it was chosen to be the dissolution medium.
3.2. In vitro drug release
It was reported that there are several main effect factors in relationship with the drug release of the double-layer osmotic pump tablets,such as the suspending agent,penetration enhancer and thickness of coating[21].
3.2.1. Effect of drug-layer properties on drug release profile
3.2.1.1. Effect of categories and amount of PEO in drug-layer on drug release profile Under the condition of the other factors unchanged,chose three different molecule weight PEO in drug layer, namely 10 thousand (N10), 20 thousand (N80) and 100 thousand(N12K)to prepare the actarit double-layerd osmotic pump tablets.As it was shown in Fig.1A,the f2values obtained were 68.54(N10 vs.N80),67.25(N80 vs.N12K)and 78.45(N12K vs.N10).So the molecule weight of PEO had no marked effect on the release of actarit.However,the drug release with N10 was incompletely and dreged easily.As for N80 and N12K,in the late period of the drug release,the preparation containing N80 was slower than the preparation containing N12K,which indicated N80, namely the low molecule weight PEO cannot form the uniform drug suspension and slowed down the drug release [22,23]. And in the process of the experiment, when added the N12K into the formulation, sticking became the serious problem. So it was decided to select N80 as the suspending agent.
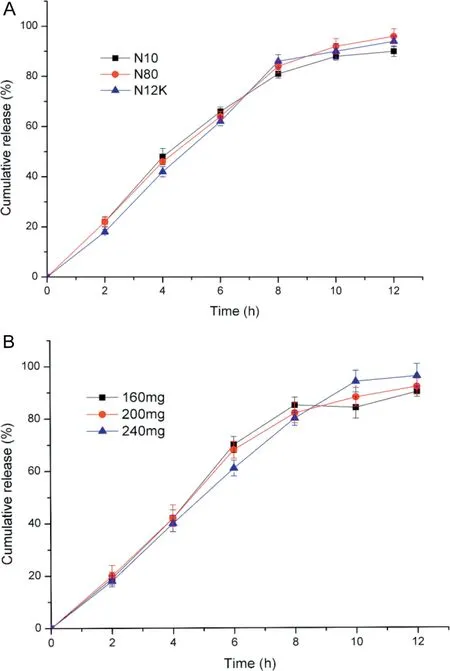
Fig.1-Effect of categories(A)and amount(B)of PEO in drug-layer on drug release profile(Mean±SD,n=6).
The amount of the N80 (160 mg,200 mg,240 mg) had been considered. The results could be seen in Fig. 1B. And the f2values obtained were 85.28 (160 mg vs.200 mg),61.79 (200 mg vs. 240 mg) and 65.84 (240 mg vs. 160 mg). It could be concluded that no significant effect existed among the different amounts.However,by contrast,cumulative release of 240 mg was nearly complete.It might be explained by the increase of PEO of drug layer increasing the viscosity of drug suspension,which increased the stability,or inhibited the aggregation and precipitation of actarit powder in suspension [24]. So 240 mg was chosen to be the final amount.
3.2.1.2. Effects of the types and amount of penetration enhancers in drug layer on drug release profile Penetration enhancers were essential to the osmotic pump tablets, so we aimed to select the best one from NaCl, lactose and mannitol. It was shown in Fig. 2A, the f2values were 71.51 (lactose vs. NaCl), 53.83 (NaCl vs. mannitol) and 51.23 (mannitol vs.lactose). Therefore, it could be concluded that no significant difference existed among the three.However,from the point of view of drug release,the cumulative release of lactose and mannitol were lower than NaCl at every pre-determined sample time.Besides,NaCl was cheaper,so NaCl was the optimal penetration enhancer.

Fig.2-Effects of the types(A)and amount(B)of penetration enhancers in drug layer on drug release profile(Mean±SD,n=6).
To investigate the influence of NaCl amount on drug release, prepared the actarit osmotic pump tablets containing distinct amount (20 mg, 50 mg, 80 mg). As illustrated in Fig. 2B, the f2values obtained were 49.16 (20 mg vs. 50 mg),48.76(50 mg vs.80 mg)and 34.84(80 mg vs.20 mg).There was evident discrepancy between the three levels.An increase in the amount of NaCl simultaneously led to an increase in the release rate of actarit[25].The release of 20 mg NaCl containing tablets was lower, whereas the two other prescriptions were similar.

Fig.3-Effect of drug loading in drug-layer on drug release profile(Mean±SD,n=6).
3.2.1.3. Effect of drug loading in drug-layer on drug release profile To investigate the influence of drug-loading on drug release,changed the dosage of actarit (120 mg,150 mg,180 mg)to compress into different tablets.The results were shown in Fig.3.The release behavior of tablets containing 120 mg drug were resemble to those of 150 mg actarit,because the f2value was 81.57 (120 mg vs. 150 mg). It can be interpreted that the increase of the drug loading could be considered as the increase of the drug concentration, because the total amount of PEO and NaCl decreased in drug layer.And the amount of released drug should also increase. However, the cumulative drug release percentage would not vary. We could also infer that the drug loading had little influence on the rheology character of drug suspension[23].Nevertheless,when the dosage increased to the 180 mg,both the release rate and cumulative release were dropped substantially. The f2values were 49.95(150 mg vs. 180 mg) and 46.33 (180 mg vs. 120 mg). It may be induced by insufficient suspension of PEO when the loading dosage was too large.
3.2.2. Effect of push-layer properties on drug release profile
The drug can be released from the double osmotic pump tablets primarily contributed to the polymer in the push layer,and its property of watering swelling can push the suspension out through the orifice [26].In order to make the release rate uniform and stable,the polymer chosen should have the excellent water swelling.Suitable permeate active substance can also promote the polymer to absorb water at the same time. In the process of researching the prescription of push layer,kept the drug layer prescription and coating membrane unchanged.
3.2.2.1. Effect of categories and amount of PEO in push-layer on drug release profile The effect of different molecular weight PEO on drug release were shown in Fig. 4A. Three kinds PEO were under consideration,WSR303(700 thousand),Coagulant(600 thousand) and WSR301 (500 thousand). The f2values of the three drug release curve were compared with each other.They were 70.72 (WSR Coagulant vs. WSR 301), 81.49 (WSR 301 vs. WSR 303) and 63.32 (WSR 303 vs. WSR Coagulant), respectively. The results indicated that variety of PEO did little effect on the release.While WSR303 was discovered that it was well pressed, and achieved the best watering rate above the three during the whole experiment.Therefore,after the comprehensive consideration,WSR303 should be the best polymer in the push-layer.

Fig.4-Effect of categories(A)and amount(B)of PEO in push-layer on drug release profile(Mean±SD,n=6).
WSR303 had been determined to use in the prescription,and then in order to explore the whether the action of different amount of WSR303 differed from each other, 100 mg,120 mg and 140 mg had been added into the formulation,respectively. Compared the f2values of the three drug release curve with each other,the f2values obtained were 79.79(100 mg vs.120 mg),72.92(120 mg vs.140 mg)and 72.77(140 mg vs.100 mg),respectively.It can be concluded that the amount of WSR303 did not have the notable effect on the drug release.Besides, from Fig. 4B, the above conclusion could also be obtained through intuitive feeling. But some small differences still existed,with the increase of the amount of WRS303,the cumulative release increased slightly. Because polyethylene oxide with a high molecular weight in the push layer acts as a swelling agent.It is among various hydrophilic polymers that,in presence of water,control the release of the active moiety either by swelling or by swelling/erosion by forming a hydrogel[27].Until up to the 140 mg,actarit can be released completely,140 mg was therefore the most suitable amount of WSR303.
3.2.2.2. Effects of the types and amount of penetration enhancers in push layer on drug release profile NaCl,lactose and mannitol were selected as the variables to find the best penetration enhancer in push-layer.As it was shown in Fig.5A,f2values were 61.19(lactose vs.NaCl),67.00(NaCl vs.mannitol)and 72.06 (mannitol vs. lactose), respectively. It was obvious that the difference among the three was nearly negligible.By the contrast,NaCl was a better selection with more complete release.
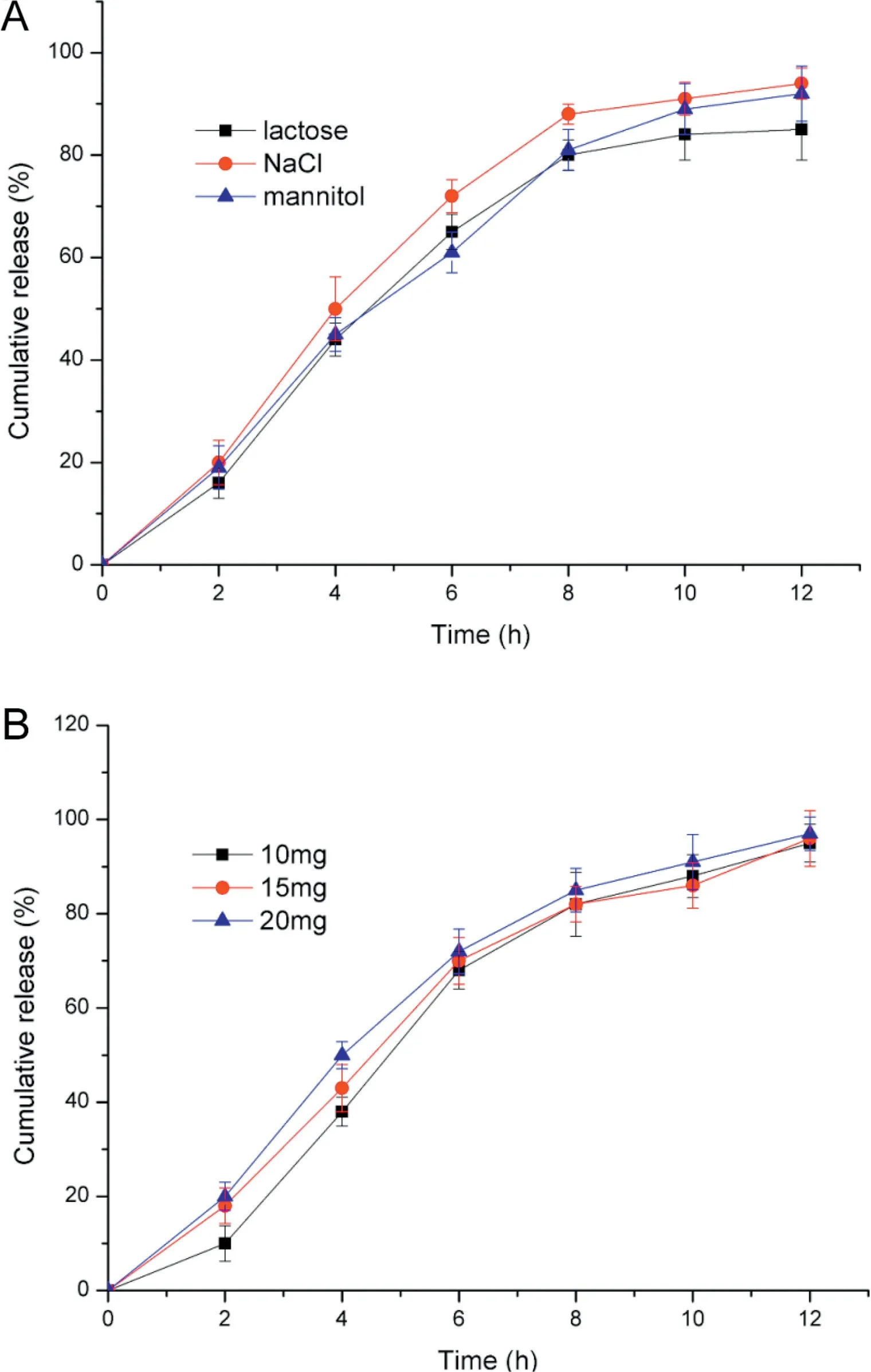
Fig.5-Effects of the types(A)and amount(B)of penetration enhancers in push layer on drug release profile(mean±SD,n=6).
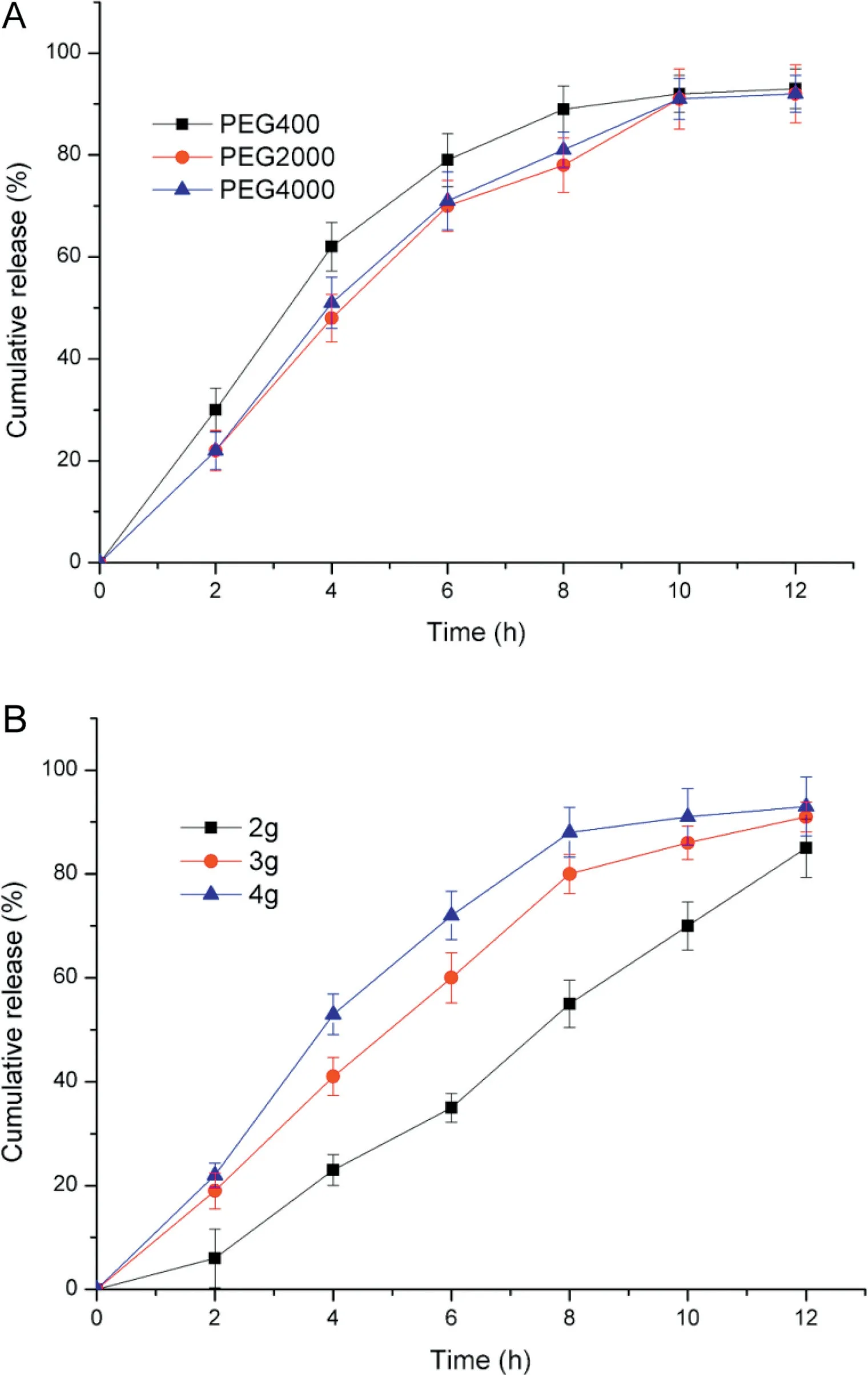
Fig.6-Effect of kinds(A)and amount(B)of pore former in coating film on drug release profile(Mean±SD,n=6).
NaCl had been employed to be the most appropriate enhancer which can be beneficial to penetration,but the amount of NaCl was under consideration. As it was indicated in Fig. 5B, even though the amount was changed (10 mg, 15 mg and 20 mg), the release patterns were similar. It also can be seen from the f2values, 72.08 (10 mg vs.15 mg), 68.82 (15 mg vs.20 mg)and 58.10(20 mg vs.10 mg).But the initial hydration rate was slightly different,a decrease in the amount of NaCl resulted in a reduced hydration rate,along with the lag-time.While the release of the tablets made of 20 mg NaCl as penetration enhancer were nearly complete.
3.2.3. Effect of properties of semi-permeable coating film on drug release profile
3.2.3.1. Effect of kinds and amount of pore former in coating film on drug release profile As is known to all, pore former was closely connected to the drug release. Plasticizers are added to modify the physical properties and improve film-forming characteristics of polymers. The role of PEG in the membrane has been described in literature with a dual functionality of plasticizer and pore former [27]. And the effect of it had been displayed in Fig. 6A. The variables were PEG400, PEG2000 and PEG4000. Significant difference existed between the different kinds of polyethylene glycol.The effects of PEG2000 and PEG4000 were nearly equal, because f2value was 80.44 (PEG2000 vs.PEG4000).However,PEG400 was faster than PEG2000 and PEG4000, the f2values of them were 47.87(PEG2000 vs.PEG400) and 52.09 (PEG4000 vs.PEG400).It could be inferred that the low molecular weight polyethylene glycol, such as PEG400 in the coating film would dissolve completely only to realize the pore forming but no plasticizing effects.But the high molecular weight polyethylene glycol may be multifunctional, one part would dissolves contributing to the pore forming,another part would be still cross-linked with cellulose acetate molecules and stay in the clothing film, resulting in the increase of toughness and strength,which met the demand of osmotic pump tablets as a coating film[28-31].PEG4000 was adopted in the following studies.
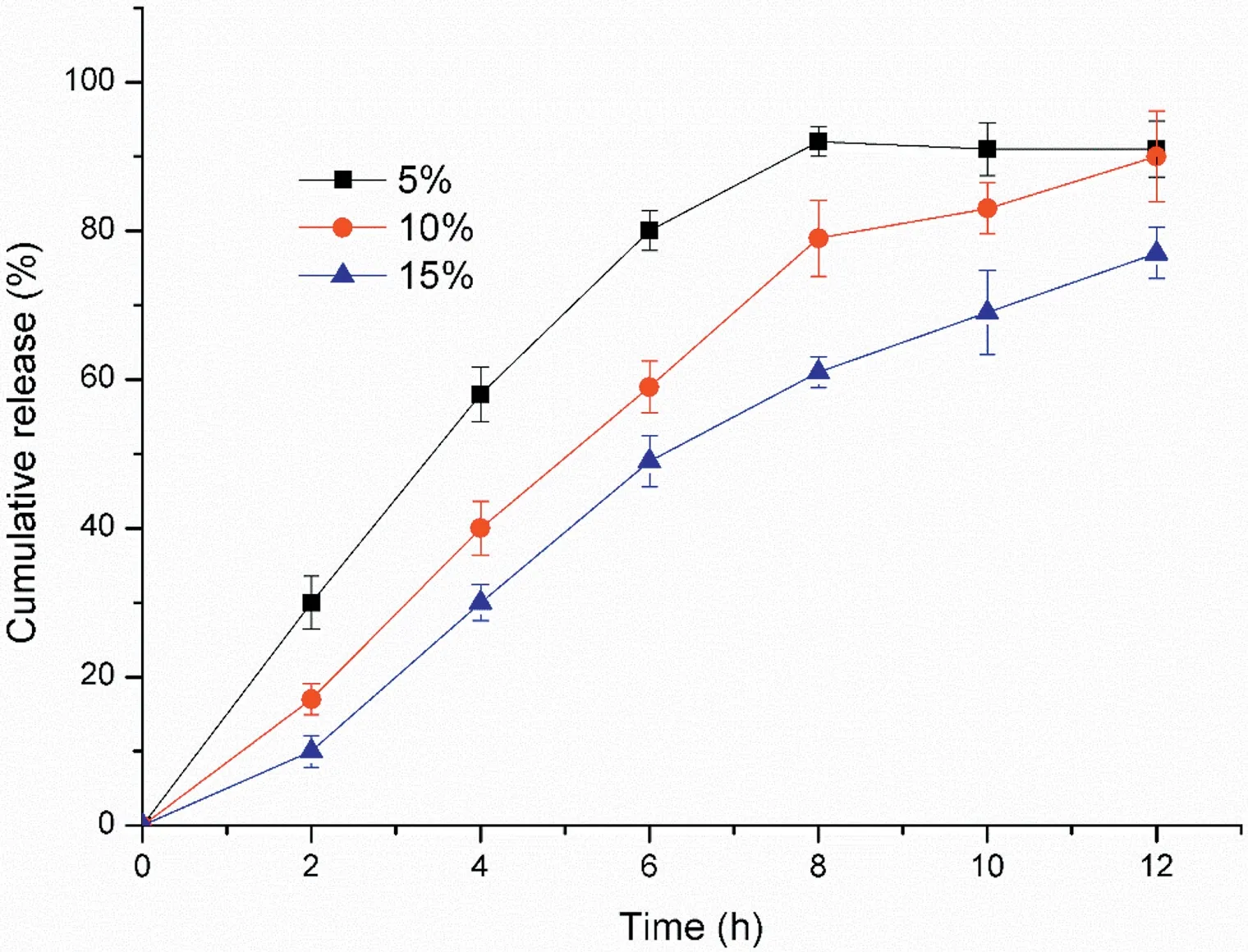
Fig.7-Effect of coating weight gain on drug release profile.(Mean±SD,n=6).
After selected the proper pore former PEG4000, then it was turn to inspect the quantity (2 g, 3 g and 4 g dissolved in 500 ml coating solution containing 3%cellulose acetate)of PEG whether affect markedly on the drug release or not. Fig. 6B told us that the release rate of actarit was proportional to the amount of PEG4000. It could be concluded that the drug was driven out faster at an ascending release rate if a larger number of PEG were added into the coating solution resulting in more pores [32].But it can be estimated from Fig.6B that the difference would decrease even disappear when the amount of pore former up to certain level.As for the f2values,we can also have such conclusions above.They were 35.47(2 g vs.3 g),56.38(3 g vs.4 g)and 28.77(4 g vs.2 g).
3.2.3.2. Effect of coating weight gain on drug release profile
The formulation of coating film almost fixed,except the coating weight gain. As it is known to all, coating weight gain is the very important factor to the osmotic pump tablets [33].Fig. 7 showed the effect of coating gain on the drug release,the levels were 5%,10% and 15%.From the point of f2values,they were 46.08 (5% vs. 10%), 43.40 (10% vs. 15%) and 31.11(15% vs. 5%). We can conclude that there was marked effect on the drug release.In this range,an increase coating gain led to a decrease drug release. In general, as the coating weight increased,the resistance of the membrane to water diffusion increased and the rate of imbibing water decreased [34].The above circumstances finally caused inadequate driving force,especially in the late release period.
3.2.4. Effect of dissolution conditions and preparation process on drug release profile
Through the confirmation of the experiment,the dissolution conditions such as rotating speed, basket method or paddle method, dissolution medium versus the preparation process such as diameter of orifice and hardness of tablets core were of no different impact on the drug release pattern[35].
3.2.5. Prescription screening and optimization
On the basis of single factor study, it was obvious that the amount of NaCl in the drug-layer, the dosage of PEG4000 in the coating solution and coating weight gain had the significant impact on the drug release.Three factor-three level was demanded in the orthogonal design to optimize the prescription, as shown in Table 2. In this study, the total score wascalculated through the weighted sum of the drug release of five pre-determined intervals,2,4,6,8 and 12 h,adopting the comprehensive scoring method. Finally, the results obtained were analyzed by intuitive analysis method. The weight of each point was set to 1,with L as the evaluation index,

Table 2-Factors and levels of orthogonal test.
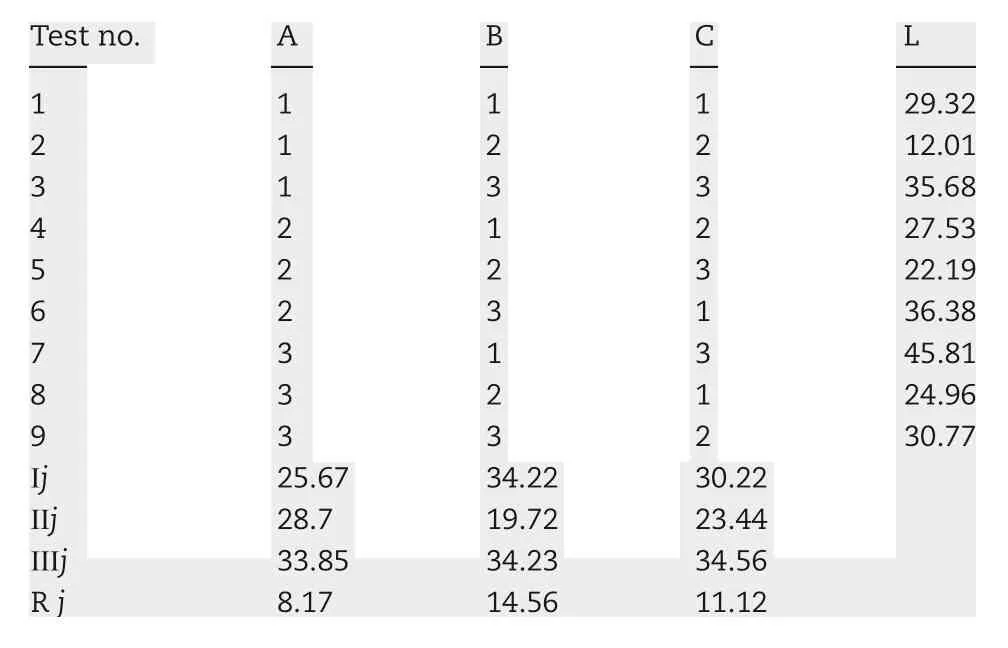
Table 3-The results of orthogonal test.
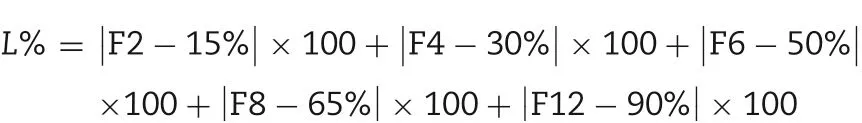
The smaller the L value is,the better the level of the factor.The results were shown in Table 3. Ⅰj, Ⅱjand Ⅲjwere corresponding to the average of one,two,three value.Rjwas the difference between the average L value of each factor.The range calculations showed that A3B2C2 was the optimal level combination of relevant factors,and the order of importance among the three factors was the dosage of PEG4000 in the coating solution, coating weight gain and the amount of NaCl in the drug-layer.
3.3. In vivo pharmacokinetic study in beagle dogs
The plasma concentration-time curves of the self-made actarit osmotic pump tablets as test tablets and commercial common tablets as reference tablets after administration in beagle dogs was presented in Fig.8.And the concentration parameters of the test and reference tablets were respectively shown in Tables 4 and 5. The Tmaxof the test tablets was 4.0 h vs.the reference tablets was 2.08 h,which indicated that the Tmaxof self-made osmotic pump tablets prolonged nearly 2 times than the commercial tablets. Great difference existed between the two according to nonparametric rank sum test (P=0.05).The average Cmaxof the self-made tablets was 1.85 μg/ml,while the commercial tablet was 2.69 μg/ml.There were significant difference between them (P <0.05) adopted one-way ANOVA test.Moreover,the AUC0-tof test tablets and reference tablets were 12.39 and 8.78 μg/(ml h), respectively.Due to the inconsistent dosage of the two preparations, the AUC0-tof self-made tablets were divided by 1.5 and then compared with commercial tablets.On the basis of the statistical analysis carried out using one-way ANOVA test,there was no significant difference between the two formulations regard to AUC0-t(P <0.05).After importing the concentration data into the DAS2.0 software, the relative bioavailability of self-made tablets and commercial tablets was 95.2%,indicating that they were bioequivalent.

Fig.8-Comparison of mean plasma concentration-time curve of actarit osmotic pump tablets with commercial tablets(Mean±SD,n=6).

Table 4-Pharmacokinetic parameters of test tablets after single dose.

Table 5-Pharmacokinetic parameters of 参考文献 tablets after single dose.
3.4. In vivo-in vitro correlation
It was obvious that the pharmacokinetics of the actarit displayed characteristics of two-compartment model after the treatment of the plasma concentration belonging to the selfmade osmotic pump tablets and commercial tablets, respectively.The absorption data in different times were calculated by Loo-Riegelman method.For the self-made tablets,the absorption data in vivo(Fa)was selected as the independent variable and the cumulative release data in vitro(Ft)as the dependent variable.The least squares linear regression was adopted.The linear equation was y=0.8575x-8.2109(r=0.9945).It suggested that good correlation existed between the in vivo absorption and the in vitro drug release. Consequently, the in vivo absorption could be well predicted by the in vitro drug release.
4. Conclusions
Actarit osmotic pump tablets were successfully prepared in this study to overcome the weak point of short half-life and greatly concentration fluctuation of actarit.Orthogonal design was applied for the optimal prescription on the basis of singlefactor inspection of drug-layer, push-layer and coating film,etc.The formulation determined finally achieved the desired effect which can realize the constant drug release rate at the first 10 h. Besides, according to the in vivo pharmacokinetic study in beagle dogs, the Tmaxof self-made osmotic pump tablets were almost 2-fold than that of commercial common tablets.And at the same time,both the bioavailability of them were approximately equal.Moreover,the in vivo drug absorption could be predicted by in vitro drug release with outstanding in vivo-in vitro correlation.Above all,the design of the actarit osmotic pump tablets was feasible.
Conflict of interest
The authors report no conflicts of interest.The authors alone are responsible for the content and writing of this article.
猜你喜欢
国际比较文学(中英文)(2020年3期)2020-11-17
肾脏病与透析肾移植杂志(2020年1期)2020-03-23
Defence Technology(2019年5期)2019-11-18
国际比较文学(中英文)(2019年1期)2019-11-12
国际比较文学(中英文)(2019年1期)2019-11-12
国际比较文学(中英文)(2018年1期)2018-11-13
转化医学杂志(2018年2期)2018-04-23
东方教育(2016年4期)2016-12-14
 Asian Journal of Pharmacentical Sciences2019年3期
Asian Journal of Pharmacentical Sciences2019年3期
- Asian Journal of Pharmacentical Sciences的其它文章
- Recent strategies on targeted delivery of thrombolytics
- Cellulose based polymers in development of amorphous solid dispersions
- Phyto-phospholipid complexes(phytosomes):A novel strategy to improve the bioavailability of active constituents
- MSNCs and MgO-MSNCs as drug delivery systems to control the adsorption kinetics and release rate of indometacin
- Effects of granulation process variables on the physical properties of dosage forms by combination of experimental design and principal component analysis
- Percutaneous absorption and brain distribution facilitation of borneol on tetramethylpyrazine in a microemulsion-based transdermal therapeutic system