
Mn掺杂锶铁氧体SrFe12O19电子结构及磁性的第一性原理研究
2019-12-24 09:26王中查显弧吴泽黄庆都时禹
无机材料学报 2019年10期
王中, 查显弧, 吴泽, 黄庆, 都时禹
Mn掺杂锶铁氧体SrFe12O19电子结构及磁性的第一性原理研究
王中1,2, 查显弧2, 吴泽1, 黄庆2, 都时禹2
(1. 哈尔滨理工大学 材料科学与工程学院, 哈尔滨 150040; 2. 中国科学院 宁波材料技术与工程研究所, 先进能源材料工程实验室, 宁波 315210)
为了揭示掺杂离子对具有磁铅石构型的锶铁氧体材料磁性能的影响, 本研究探讨了锶铁氧体及其锰掺杂体系的稳定构型及其磁结构。研究结果表明, 锶铁氧体为亚铁磁性, 与前期的研究结果相吻合。通过比较GGA和GGA+U计算方法, 发现U值的选取对体系的电子结构和原子磁矩有显著影响。当U值为3.7 eV时, 体系由金属性转变为自旋向上带隙为1.71 eV的半导体。原胞总磁矩为40 μB。对于Mn替换掺杂的SrFe12–xMnO19体系, 通过不同占据位能量比较, 当单个Mn原子替换(=0.5)时, Mn离子优先占据Fe(12k)位置; 而当两个Mn原子替换Fe原子(=1.0)时, 两个Mn分别占据Fe (12k)和Fe (2a)位置。Mn掺杂对锶铁氧体的结构影响较小, 但对于体系的总磁矩和电子结构有较明显的影响。在Mn含量=0.5和=1.0时, 自旋向上带隙值分别降低到0.85和0.59 eV, 原胞的总磁矩为39和38 μB。本研究可为实验研究提供理论指导。
锶铁氧体; Mn掺杂; 第一性原理; 电子结构; 磁矩
磁性材料在仪器仪表、通信设备、电动机、传感器等多个领域都有重要应用[1-5]。铁氧体作为永磁材料的重要组成部分, 其原材料丰富、工艺简单、成本低, 获得了工业界的广泛青睐。铁氧体主要分为锶铁氧体和钡铁氧体, 其中锶铁氧体的各向异性常数和矫顽力相对于钡铁氧体更高, 并且重量更轻, 易于满足生产领域对高档永磁铁氧体的需求[6-8]。相对于含稀土金属的Nd-Fe-B磁性材料, 锶铁氧体的矫顽力仍需进一步提高, 结构较脆。因此, 为了获得更高性能的锶铁氧体材料, 实验和工业上常采用离子掺杂的方式来改善材料的性能。前期研究表明, 采用Ce[9]、La[10-11]、Sm[12]、Pr[13]、Nd[14]替换单个Fe离子, 增加了锶铁氧体的矫顽力, 对于Zr-Cd[15], Er-Ni[16]、Sn-Mg[17]、Co-Nd[18]、Mg-Ti[19]共掺杂替换Fe离子, 改善了锶铁氧体的饱和磁化强度。在理论方面, Vivek等[20]采用第一性原理方法研究了Al掺杂锶铁氧体对其磁性的影响, 确定不同Al含量替换的稳定构型, Al掺杂同样提高了锶铁氧体的矫顽力。Liyanage等[21]研究了Zn-Sn共掺杂对锶铁氧体磁性能的影响, 获得了更高饱和磁化强度的掺杂体系。尽管已有大量的掺杂研究工作, 但是对于Fe的邻近元素Mn掺杂锶铁氧体的尚未见相关报道。
我国具有丰富的Mn资源, 制备工艺成熟。同时Mn是Fe的邻近元素, 在掺杂工艺上更易实现。此外, 从原子层电子排布来说, Mn原子的最外层电子排布为3d54s2, 而Fe离子的最外层电子排布为3d64s2, Mn较Fe具有更高的电子自旋磁矩。因此, 通过Mn掺杂锶铁氧体有望获得制备工艺简单、成本低、性能优的锶铁氧体体系。本研究采用第一性原理探讨了锶铁氧体的稳定磁结构以及计算方法的影响, 并进一步考察了不同含量Mn掺杂锶铁氧体的稳定构型、电子结构以及磁性能, 有望为未来实验室制备性能优良的锶铁氧体材料提供理论依据。
1 计算方法
基于第一性原理密度泛函理论, 本研究所有计算工作都在平面波软件VASP (Viennasimulation Package)[22-23]中进行。交换关联泛函选取GGA (Generalized Gradient Approximation)近似下的PBE (Perdew-Burke-Ernzerho)形式[24], 势函数选用投影缀加波PAW (Projector augmented-wave)势[25-26], 平面波截断能设为520 eV。对于锶铁氧体以及Mn掺杂体系, 选取SrFe12O19晶胞作为研究体系。对于晶格优化, 布里渊区内K点采用Monkhorst-Pach方式的7´7´1的网格[27]。所有结构充分弛豫到每个原子的受力小于0.01 eV/Å (1 Å=0.1 nm)。由于考察的结构含有3d过渡金属Fe和Mn元素, 其电子间相互作用较强, GGA一般低估体系能带带隙和晶格参数, 本研究考察了GGA+U方法对体系进行修正。对于Fe和Mn的3d电子层, 基于前期文献报道的结果, U值分别选取为3.7[28]和 3.5 eV[29-30]。
2 结果与讨论
2.1 未掺杂锶铁氧体构型
在考察Mn掺杂体系之前, 首先考察锶铁氧体的结构及稳定的磁结构分布。锶铁氧体为六角晶格(图1), 空间群为P63/MMC。晶胞结构中具有64个原子, 包含2个Sr原子, 24个Fe原子以及38个O原子。根据原子等效占位的不同, Fe的原子位置可分为2a、2b、4f1、4f2、12k等五种不同占位。根据不同位置Fe原子磁矩方向的不同会形成铁磁以及多种亚铁磁构型。根据排列组合关系, 我们考察了9种可能的铁磁和亚铁磁构型, 具体构型如表1所示。其中“+”表示磁矩方向向上, “-”表示磁矩方向向下。通过结构弛豫不同的磁结构发现, 2a、2b以及12k位置的Fe原子磁矩向上, 4f1和4f2处Fe原子磁矩向下的亚铁磁构型具有最低的能量, 表明这种亚铁磁构型是锶铁氧体的稳定态。此计算结果与前期的文献报道相吻合[31], 表明计算结果可靠。因此, 后续的电子结构、磁性能以及掺杂体系都将基于这种稳定的亚铁磁构型进行计算。

图1 锶铁氧体晶胞结构
(a) The green ball denotes the Sr atom, the small grey ball denotes the O atom; (b) The blue, red, purple, magenta, and blue-green balls represent the Fe atoms in 12k, 4f2, 4f1, 2a, and 2b sites, respectively. The spin directions for different Fe3+are labeled with different colors (black and red)
值得注意的是, 前述采用GGA方法比较了不同铁磁和亚铁磁结构分布, 获得了锶铁氧体稳定磁结构。但由于Fe原子含有高简并度的d电子轨道, 电子间的自相互作用较强, 需要考虑U值进行修正。基于之前文献的报道结果[28], 我们对Fe原子的d轨道考虑U=3.7 eV的修正。研究结果表明, 稳定的磁结构与GGA的计算结果相吻合, 但是结构和电子性能存在一定差异。对于稳定的磁结构, 其晶格参数较不加U时有显著增加, 其水平面内以及垂直方向的晶格参数分别由0.586和2.310 nm增加到0.594和2.319 nm。含U值计算得到的晶胞参数与实验测量值=0.588 nm,=2.304 nm更吻合[32], 表明加U计算对于锶铁氧体体系是必要的。此外, 表格2中给出了基于两种方法得到具体的原子磁矩。不加U时, 不同位置Fe原子磁矩在3.17~3.73 μB。加U后, 单个原子磁矩有较大幅度的增加, 磁矩范围为4.05~4.18 μB, 其中12k和2a位置磁矩略高于其他位置, 晶胞的总磁矩为40 μB。对于电子结构的研究, 图2分别给出了锶铁氧体不加U和加U条件下的电子态密度和能带结构。不加U情况下, 能带结构和态密度如图2(a~c)所示, 可以看出锶铁氧体呈现金属性, 与实验值测得的高电阻率明显不符[33]。加U后, 锶铁氧体呈现半导体特性。从态密度图2(d)中可以看出, 对于占据态来说, 12k、2a、2b具有较大的自旋向上电子态, 而4f1和4f2具有较大的自旋向下电子态, 这与锶铁氧体亚铁磁构型相吻合。12k和2b处Fe原子以及O原子态密度更接近体系的价带顶, 而导带底不同占据位的Fe原子以及O原子的态密度比较接近。整个能量区间内, Fe和O原子的态密度有较大的重叠区间, 表明Fe和O原子之间有较强的轨道杂化和成键。通过加U计算, 获得的能带图如图2 (b), 自旋向上带隙值为1.71 eV, 自旋向下带隙值为1.82 eV, 与实验测得的高电阻率较为符合。通过比较加U和不加U条件下晶格参数和电子结构的计算, 发现加U计算与实验结果更接近。所以后续掺杂体系的研究都采用GGA+U的方法。
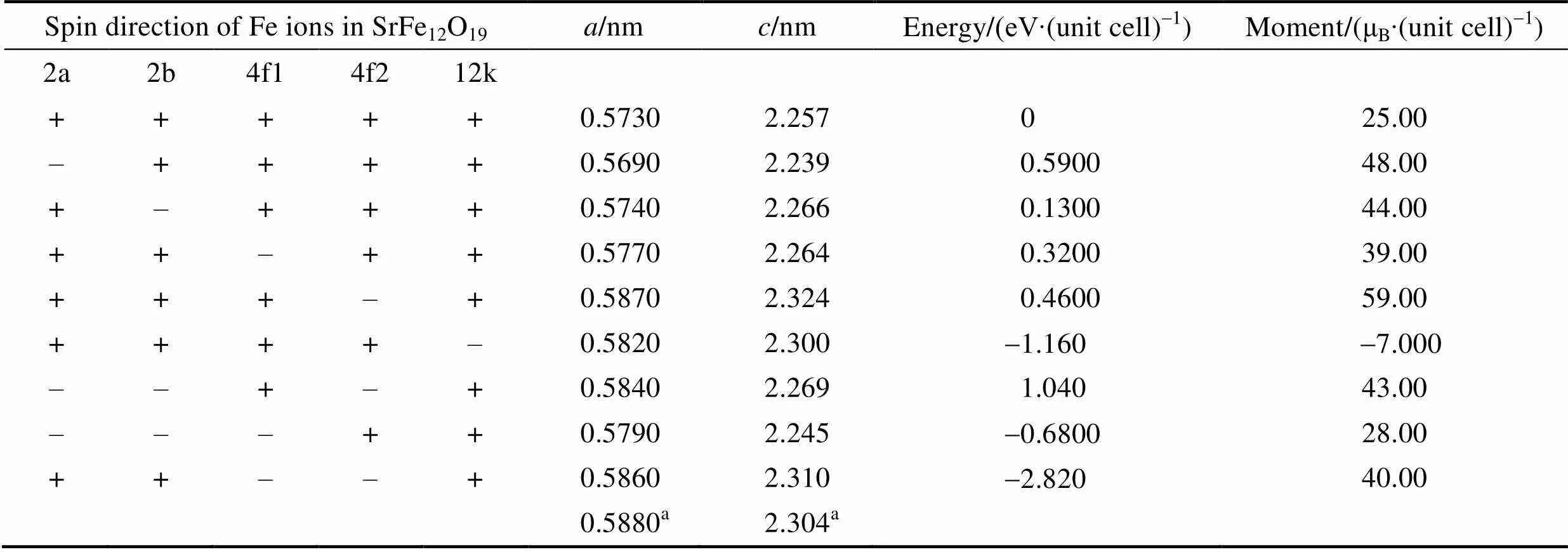
表1 不同磁构型锶铁氧体的晶格常数、相对能量和净磁矩
a: exp. Ref.[32]
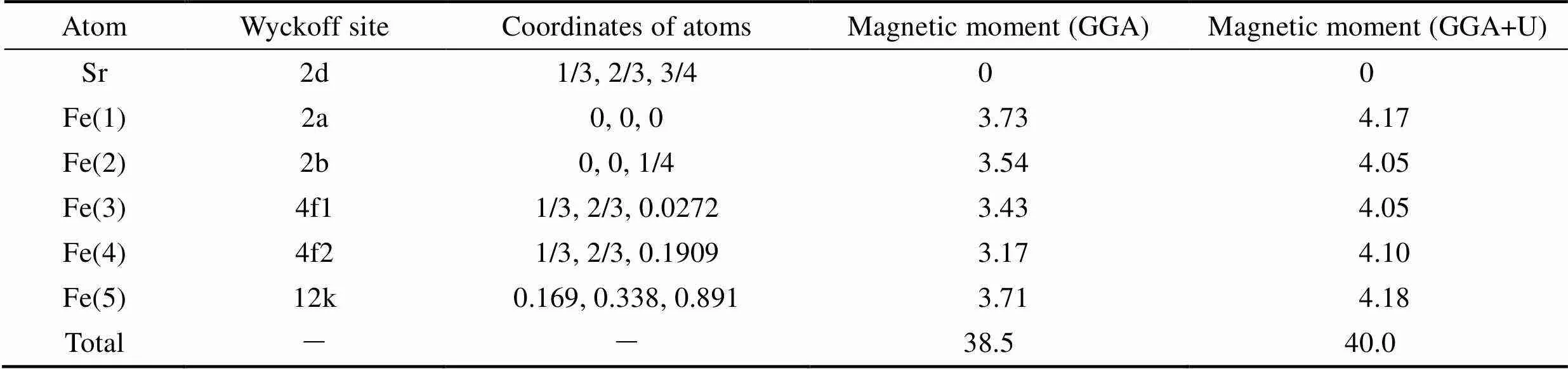
表2 GGA和GGA+U条件下锶铁氧体的Fe原子磁矩及晶胞总磁矩(μB)
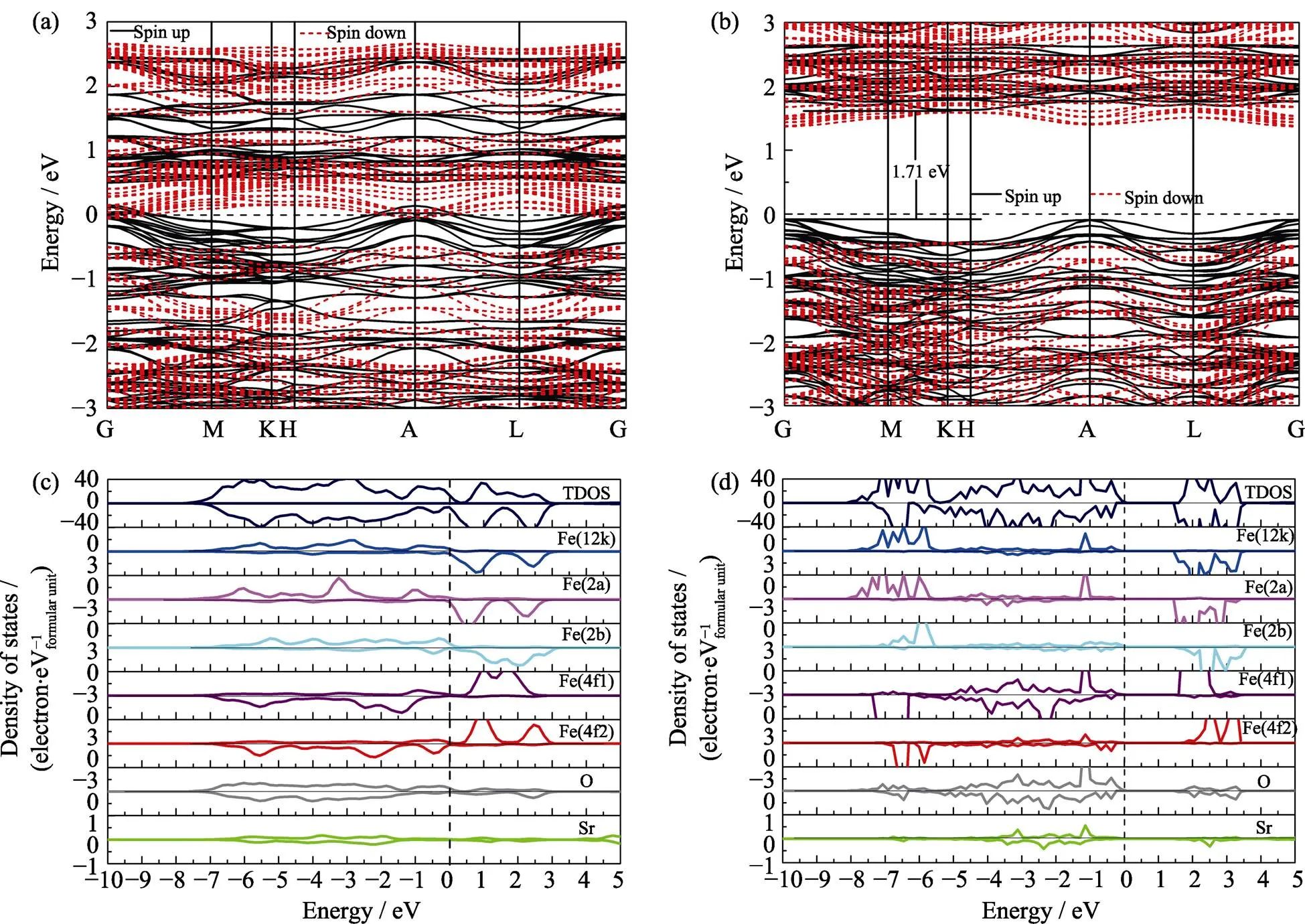
图2 基于GGA计算的锶铁氧体的电子能带结构图(a)和总态密度、原子分态密度图(c), 基于GGA+U计算的锶铁氧体的电子能带结构图(b)和总态密度、原子分态密度图(d)
2.2 Mn掺杂锶铁氧体SrFe12–xMnxO19对磁性及电子结构的影响
基于对锶铁氧体计算方法和稳定磁结构的讨论, 我们进一步采用GGA+U方法考察Mn掺杂锶铁氧体的本征物性。掺杂体系采用SrFe12–xMnO19表示。
基于文献[20]的报道, 我们考察了(=0.5, 1.0)两种低含量掺杂构型。基于锶铁氧体的晶胞结构,=0.5表示一个Mn原子在锶铁氧体晶胞中替换Fe原子;=1.0表示两个Mn原子替换锶铁氧体晶胞中两个Fe原子。为了确定Mn原子是否可替换Fe以及更容易替换哪种类型的Fe原子, 根据公式(1)我们对替代能进行了计算:

公式中(())表示Mn原子取代Fe后掺杂锶铁氧体的总能量,()表示未掺杂锶铁氧体晶胞的总能量, Δ()表示掺杂原子和原有原子在各自最稳定晶格中的能量差(= Fe, Mn),表示原子增加或减少的数量, 表3列出了计算结果。
从表3中可以看出, 单个Mn原子替代体系在不同取代位的能量差别较大。如在12k位置, 取代能能量最低, 为–2.50 eV, 表明单个Mn原子替换, 首先取代12k位置的Fe原子; 在2a和4f2位置取代能量分别为–2.24和–2.03 eV, 略高于12k位置。但是随着Mn含量的增加以及温度升高等效应, 这两个位置的Fe原子仍有一定可能被取代。由于2b位置的原子与Sr原子间距较小, 所以原子之间的相互作用较强。但是从理论计算发现, Mn替换2b位置的Fe离子取代能最高, 为–0.34 eV, 说明2b位置的Fe与Sr的相互作用要比Mn与Sr更强。因此Mn原子与Sr原子之间存在较少的电子对, 导致取代Fe离子后磁矩增大。总体而言, 所有取代位的能量都为负值, 表明通过Mn掺杂取代在实验上易于实现, 只是不同位置Fe原子被取代的可能性存在差异。对于Mn取代后的体系的磁矩而言, 单个Mn原子取代对体系,相对于Al原子的取代情形[20], 磁矩的变化量相对较小, 如2a、12k和4f1位置的取代晶胞磁矩减小1 μB, 而4f2位置取代晶胞磁矩增加1 μB。虽然2b位置取代磁矩增加较显著, 但其取代能较高, 被取代的可能性较低。因此, 可以看出, 通过Mn掺杂取代能较好地保持锶铁氧体的磁化强度。
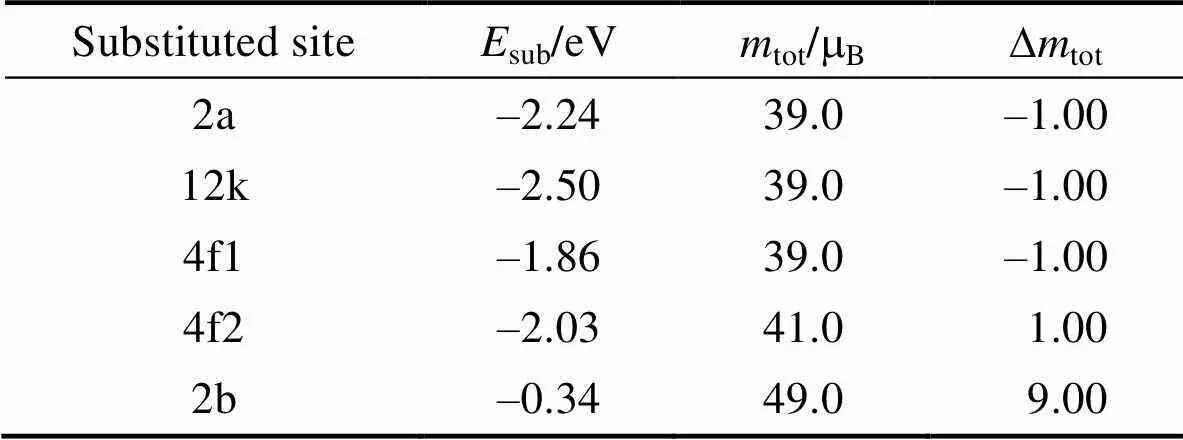
表3 单个Mn原子替代掺杂锶铁氧体不同位置Fe原子的替代能Esub、替代后的晶胞总磁矩mtot和磁矩的改变大小Dmtot
两个Mn原子取代的SrFe12–xMnO19(=1.0)结构如何, 本研究基于前面单个原子取代计算得到的取代能, 12k和2a位置取代能量较低, 因此研究主要考察两个Fe原子在这两种位置的取代情形。类似之前文献报导对Al替代情形的分析[20], 将不同位置取代的构型按照“[第一个Mn原子取代位, 第二个Mn原子取代位].编号”的格式进行标记。例如, 当两个Mn原子分别取代2a和12k位置时, 有两种不同的构型, 分别标记为[2a,12k].1 和[2a,12k].2。将能量较低的十种替代构型列于表格4中。从表格中 可以看出, 当两个 Mn 原子分别取代近邻的2a和12k位置 Fe原子时, 取代能量最低为–4.02 eV, 表明两个Mn原子取代时优先取代2a和12k位置的Fe原子。值得注意的是, [12k, 12k].7, [12k,12k].6以及[2a,12k].2的取代能与[2a,12k].1构型的取代能比较接近, 在实验上仍然存在一定的几率被取代。类似地, 本研究考察了掺杂后体系的磁矩变化。从表格中可以看出, 不管是最稳定的构型[2a,12k].1还是其他替代掺杂构型, 体系的磁矩都呈下降趋势, 且下降的幅度都为2mB。体系的总磁矩随着Mn含量增加而减小。

表4 两个Mn原子替换掺杂情况下, SrFe12–xMnxO19(x=1.0)不同构型的取代能Esub, 晶胞磁矩mtot, 以及相对于未掺杂体系的晶胞磁矩变化量Dmtot
为了更加深入理解掺杂体系磁矩的变化量, 我们考察了单个Mn原子替换掺杂12k和2a位置的Fe原子位, 以及两个Mn原子掺杂稳定构型[2a,12k].1中每个原子对总磁矩的贡献, 并与未掺杂体系进行对比, 具体数据如表格5所示。从表格中可以看出, 锶铁氧体的磁矩主要来源于Fe原子, 此外O原子有比较小部分的贡献。当单个Mn原子掺杂替换12k位Fe原子时, Mn比Fe呈现的原子磁矩小, 为3.798mB, 较其替代的Fe原子, 磁矩减小了0.383mB。由于Mn原子的取代, 影响了整体体系的键合作用, O原子和间隙位的磁矩也出现减小, 导致整个晶胞的磁矩减小了1mB。类似地, 当Mn取代2a位Fe原子时, Mn较其替代的Fe磁矩减小了0.299mB, 体系的总磁矩减小了1mB。在两个Mn原子取代[2a,12k].1构型中, 2a和12k位置的Mn原子较其替代的Fe原子磁矩分别减小了0.397和0.381mB, 体系的总磁矩减小2mB。
基于上面的讨论, 单个Mn优先取代12k位置的Fe原子, 而当两个Mn取代时, 优先取代近邻位置的2a和12k位置的Fe原子。随着Mn含量的增加, 体系的磁矩有较小幅度的减小。通过单个原子的磁矩的分析, 确定了磁矩变化量的根源。基于上述确定的稳定磁结构, 我们进一步探讨了Mn掺杂对体系的晶格参数以及电子结构的影响。表格6中给出了未掺杂锶铁氧体以及两种不同Mn含量掺杂的稳定构型的晶格参数和体积。与未掺杂锶铁氧体的晶格参数和体积相比, Mn掺杂后晶格常数和体积变化不明显。Mn掺杂SrFe12–xMnO19中=0.5时, 对应的平面内垂直方向上的晶格常数和分别为0.594和2.319 nm。Mn掺杂SrFe12–xMnO19中=1.0时, 对应的晶格常数和分别为0.595和2.320 nm。与未掺杂的锶铁氧体晶格常数相比, 晶格常数的变化小于0.34%。晶格和体积变化较小的原因主要是由于Mn原子的半径和Fe原子的半径比较接近, Mn取代Fe原子后, 不会引起较大的晶格畸变。
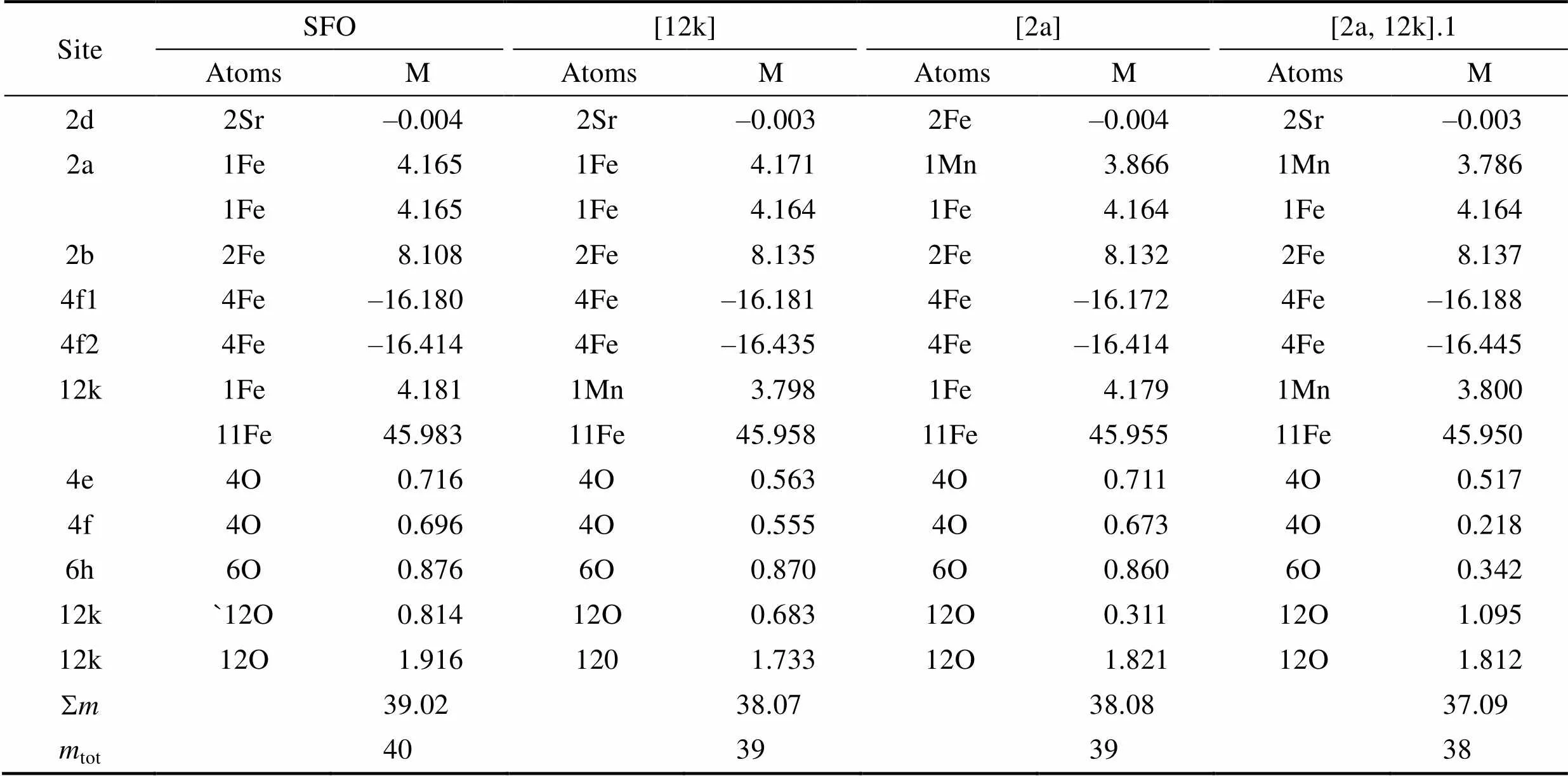
表5 锶铁氧体SFO、单个Mn替换12k位置Fe的掺杂模型[12k]、单个Mn替换2a位置Fe的掺杂模型[2a], 以及两个Mn原子掺杂稳定构型[2a,12k].1的四种不同构型中原子磁矩分布
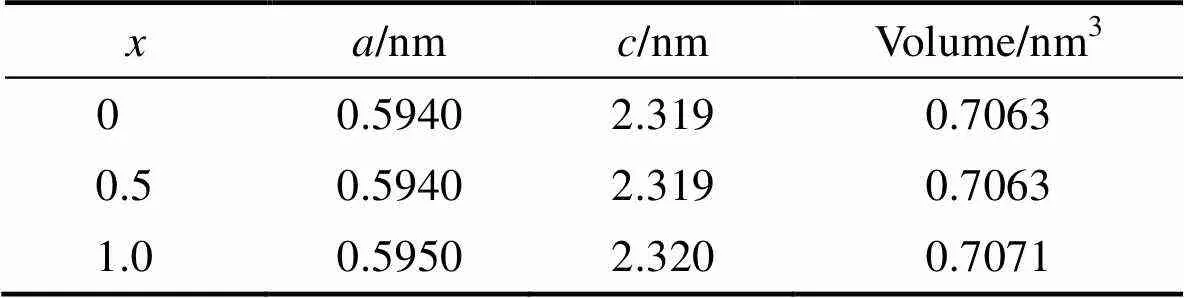
表6 未掺杂和Mn掺杂锶铁氧体的晶格常数和体积
在此基础上进一步考察Mn掺杂对体系电子结构的影响。单个Mn取代12k位置Fe原子的锶铁氧体构型的能带结构图和电子态密度图(图3(a, c)显示, Mn较Fe原子具有更高能级的占据态,构建了掺杂体系的价带顶, 导带底主要由Mn和2a, 12k位置的Fe原子轨道组成。由于价带顶的上移, 导致体系的自旋向上能带带隙显著降低, 自旋向上带隙值降低到0.85 eV, 自旋向下带隙值为1.82 eV, 带隙值的降低, 在一定程度上提升了体系的导电性能。此外, Mn与O的态密度在价带部分有较宽能量区间的重叠, 表明掺杂后Mn与周围的O原子有更强的键合作用。图3(b, d)给出了两个Mn原子掺杂情形下的稳定构型[2a. 12k].1的能带结构图和电子态密度图。整体上而言, 两个Mn掺杂的态密度与单个Mn掺杂的态密度图相似, 掺杂的Mn原子的电子态对体系的导带底和价带顶有较大贡献。体系的自旋向上带隙值进一步减小, 自旋向上带隙值为0.59 eV, 自旋向下带隙值为1.84 eV。在价带能量区间, Mn的电子态和O的电子态仍有较大区间的重叠。从替代能的计算公式中可以看出, Mn掺杂体系的能量有显著降低。但体系的晶格参数变化很小, 说明通过Mn的掺杂有望提升体系的力学性能。总体而言, Mn的掺杂降低体系的带隙, 提升锶铁氧体的导电性能, 同时有望提升体系的力学强度。
3 结论
本研究采用第一性原理密度泛函理论研究了锶铁氧体以及Mn掺杂锶铁氧体的结构、电子结构以及磁学性能。比较了GGA和GGA+U对锶铁氧体结构和电子结构的影响, 结果表明:
1) 锶铁氧体的稳定磁结构为亚铁磁性。加U对体系的晶格参数和电子结构有显著影响。通过加U理论计算的晶格常数为==0.594 nm,=2.319 nm, 与实验值符合得更好。通过对电子结构的分析, 未掺杂体系的带隙值为1.71 eV, 与实验的高电阻率相符。通过磁性的分析, 锶铁氧体具有很强的自旋极化效应, 晶胞的磁矩为40mB。
2) 通过对Mn掺杂锶铁氧体的考察发现, 单个Mn原子优先替换12k位置的铁原子; 当两个Fe原子替换时, 优先替换近邻位置的12k和2a位置。随着Mn含量的增加, 体系的磁矩有减小趋势, 如单个Mn和两个Mn替换后体系的磁矩分别减小了1和2mB。此外, Mn掺杂对体系的晶格参数影响较小, 但对电子结构有显著影响。随着Mn含量的增加, 体系的带隙值减小。基于晶格参数和电子结构的分析, 通过Mn掺杂, 有望提升体系的力学性能。本研究可为Mn掺杂锶铁氧体的实验研究提供有益的参考, 期待实验的进一步证实。
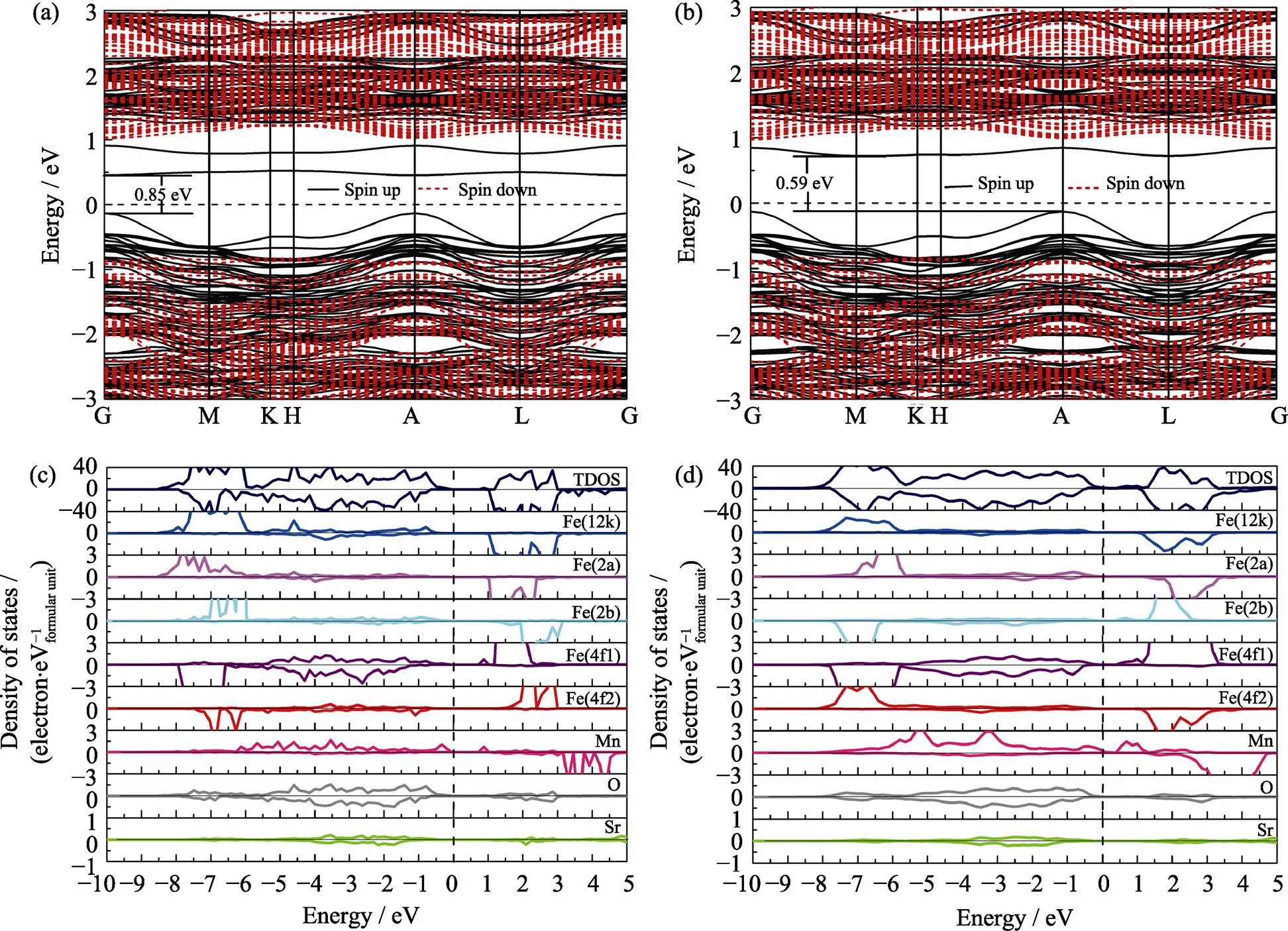
图3 基于GGA+U计算的单个Mn原子掺杂SrFe12–xMnxO19 (x=0.5)的电子能带结构图(a)和总态密度、投影态密度图(c), 基于GGA+U计算的Mn掺杂锶铁氧体(x=1.0)的电子能带结构图(b)和总态密度、投影态密度图(d)
[1] ZHOU X, MA L, LIU T,. Grystal structure and magnetic property of Si3N4/FePd/Si3N4thin films., 2018, 33(8): 909–915.
[2] MENG F B, MA X F, ZHANG W,. Structure and magnetic property of Fe and spinel Co2MnO4., 2017, 32(6): 609–614.
[3] XIAO L, CHEN Y, LIU Z,. Growth, magnetic and electrical transport properties of La0.7Sr0.3MnO3thin films on plzst ceramics., 2017, 32(3): 326–330.
[4] ZHANG L, LIU HH, LIU LJ,. Effects of La doping on CaB6thin films prepared by DC magnetron sputtering., 2017, 32(5): 555–560.
[5] ZHAO X Y, MAN P W, XIE T,. Crystal growth and characterization of the rare-earth orthoferrite Sm0.8Tb0.2FeO3single crystal., 2016, 31(9): 1004–1008.
[6] SEEMA V, JOY P A, KHOLLAM Y B,. Synthesis of nanosized MgFe2O4powders by microwave hydrothermal method., 2004, 58(6): 1092–1095.
[7] CHEN D, LIU Y, LI Y,. Microstructure and magnetic properties of Al-doped barium ferrite with sodium citrate as chelate agent., 2013, 337–338: 65–69.
[8] WANG H W, KUNG S C. Crystallization of nanosized Ni-Zn ferrites powders prepared by hydrothermal method., 2004, 270(1/2): 230–236.
[9] ALMESSIERE M A, SLIMANI Y, BAYKAL A,. Structural and magnetic properties of Ce-doped strontium hexaferrite., 2018, 44: 9000–9008.
[10] SEIFERT D, TÖPFER J, LANGENHORST F,. Synthesis and magnetic properties of La-substituted M-type Sr hexaferrites., 2009, 321(24): 4045–4051.
[11] WANG J F, PONTON C B, GRÖSSINGER R,. A study of La-substituted strontium hexaferrite by hydrothermal systhesis., 2004, 369(1/2): 170–177.
[12] WANG J F, PONTON C B, HARRIS I R,. A study of the magnetic properties of hydrothermally synthesisted Sr hexaferrite with Sm substitution., 2001, 234(2): 233–240.
[13] WANG J F, PONTON C B, HARRIS I R,. A study of Pr-substituted strontium hexaferrite by hydrothermal synthesis., 2015, 403(1/2): 104–109.
[14] WANG J F, PONTON C B, HARRIS I R,. A study of Nd-substituted Sr hexaferrite prepared by hydrothermal synthesis., 2002, 38(5): 2928–2930.
[15] ASHIQ M N, IQBAL M J, GUL I H,. Structural, magnetic and dielectric properties of Zr-Cd substituted strontium hexaferrite (SrFe12O19)., 2009, 487(1/2): 341–345.
[16] ASHIQ M N, IQBAL M J, NAJAM-UL-HQ M,. Synthesis, magnetic and dielectric properties of Er-Ni doped Sr-hexaferrite nanomaterials for applications in high density recording media and microwave divices., 2012, 324(1): 15–19.
[17] DAVOODI A, HASHEMI B. Magnetic properties of Sn-Mg substituted strontium hexaferrite nanoparticles synthesizedcoprecipitation method., 2011, 509(19): 5893–5896.
[18] CHEN W, WU W W, ZHOU C,. Structural and magnetic properties evolution Co-Nd substituted M-type hexagonal strontium ferrites synthesized by ball-milling-assisted ceramic materials., 2018, 47(3): 2110–2119.
[19] EBRAHIMI F, ASHRAFIZADEH F. Tuning the ferromagnetic resonance by doping strontium hexaferrite nanopowders., 2018, 85(3): 621–628.
[20] VIVEK D, CHANDANI N, NANDADASA,. Site occupancy and magnetic properties of Al-substituted M-type strontium hexaferrite., 2015, 17: 243904–243912.
[21] LIYANAGE L S I, KIM S, HONG Y K,. Theory of magnetic enhancement in strontium hexaferrite through Zn-Sn pair substitution., 2013(348): 75–81.
[22] KRESSE G, FURTHMÜLLER J. Efficient iterative schemes fortotal-energy calculations using a plane-wave basis set., 1996, 54(16): 11169–11186.
[23] KRESSE G, FURTHMÜLLER J. Efficiency oftotal energy calculations for metals and semiconductors using a plane-wave basis set., 1996, 6: 15–50.
[24] PERDEW J P, BURKE K, EMZERHOF M. Generalized gradient approximation made simple., 1996, 77: 3865–3868.
[25] BLÖCHL P E. Projector augmented-wave method., 1994, 50: 17953–17979.
[26] KRESSE G, JOUBERT D. From ultrasoft pseudopotentials to the projector augmented-wave method., 1999, 59: 1758–1775.
[27] MONKHORST H, PACK J. Special points for Brillouin-zone interations., 1976, 13: 5188–5192.
[28] LIYANAGE L S I, KIM S, HONG Y K,. Theory of magnetic enhancement in strontium hexaferrite through Zn-Sn pair substitution., 2013, 348: 75–81.
[29] HUANG L H, ZHU Q S, GE W,. Oxygen-vacancy formation in LaMO3(M = Ti, V, Cr, Mn, Fe, Co, Ni) calculated at both GGA and GGA + U levels., 2011, 50: 1800–1805.
[30] SAHU B R, BANERJEE S K. Density-functional study of bulk silicon lightly doped with manganese., 2008, 77(15): 155203–155209
[31] GORTER E W. Saturation magnetization of some ferromagnetic oxides with hexagonal crystal structures., 1957, 104: 255–260.
[32] KIMURA K, OHGAKI M, TANAKA K,. Study of the bipyramidal site in magnetoplumbite–like compounds SrM12O19(M=Al, Fe, Ga)., 1990, 87: 186–194.
[33] IQBAL M J, FAROOQ S. Impact of Pr-Ni substitution on the electrical and magnetic properties of chemically derived nanosized strontium-barium hexaferrites., 2010, 505: 560–567.
First-principles Study on Electronic and Magnetic Properties of Mn-doped Strontium Ferrite SrFe12O19
WANG Zhong1,2, ZHA Xian-Hu2, WU Ze1, HUANG Qing2, DU Shi-Yu2
(1. School of Material Science and Engineering, Harbin University of Science and Technology, Harbin 150040, China; 2. Engineering Laboratory of Advanced Energy Material, Material Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Science, Ningbo 315201, China)
To reveal influence of doping ions on the magnetic properties of strontium ferrite materials with magnetoplumbite configurations, we studied the stable configurations and magnetic structures of strontium ferrite with and without manganese doping. The results show that the strontium ferrite is ferrimagnetic, which is consistent with the previous reports. Comparing the GGA and GGA+U approaches, the U value exhibits a significant impact on the electronic structures and atomic magnetic moments. When 3.7 eV is adopted for U value, the system changed from a metal to a semiconductor with a spin up band gap of 1.71 eV. The total magnetic moment of the pure strontium ferrite is 40 μB. For the SrFe12–xMnO19system, the site preference of Mn substituting Fe is investigated with=0.5 and=1.0. When= 0.5, the single doping Mn atom preferentially occupies the Fe (12k) site. For=1.0, the two Mn atoms preferentially occupy the Fe (12k) and Fe (2a) sites, respectively. Doping Mn has little impact on the lattice structure of strontium ferrite, but have a significant effect on the total magnetic moments and electronic structures. When=0.5 and=1.0, the band gap values for spin up electrons reduced to 0.85 and 0.49 eV, and the total magnetic moments reduced to 39 and 38 μB, respectively. This study may provide a theoretical foundation for future experimental studies.
SrFe12O19; Mn-doped; the first principles; electronic structure; magnetic moment
TQ174
A
1000-324X(2019)10-1047-08
10.15541/jim20190003
2018-12-29;
2019-02-11
国家重点研发计划 (2016YFB0700100) National Key Research and Development Programme (2016YFB0700100)
王中(1992–), 男, 硕士研究生. E-mail: wzhong@nimte.ac.cn; 查显弧(1988–), 男, 副研究员. E-mail: zhaxianhu@nimte.ac.cn
都时禹, 研究员. E-mail: dushiyu@nimte.ac.cn; 吴泽, 副教授. E-mail: wupuqiming@163.com
猜你喜欢
佛山科学技术学院学报(自然科学版)(2022年5期)2022-10-09
物理学报(2022年15期)2022-08-12
光子学报(2022年6期)2022-07-27
军民两用技术与产品(2022年1期)2022-06-01
西北工业大学学报(2022年1期)2022-04-22
当代作家(2021年11期)2021-12-17
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
中学生数理化(高中版.高考理化)(2019年11期)2019-11-30
中学生数理化(高中版.高考理化)(2019年11期)2019-11-27
中学生数理化(高中版.高考理化)(2019年10期)2019-11-08
