
高效液相色谱–串联质谱法检测环境水体中18 种全氟化合物
2020-02-08 09:22杨文宇赵慧钟琳李支薇
化学分析计量 2020年1期
杨文宇,赵慧,钟琳,李支薇
(1.广州广电计量检测股份有限公司,广州 510656;2.中车长春轨道客车股份有限公司,长春 130062)
全氟烷基化合物(PFASs)是一类新的持久性有机污染物(POPs),因其具有极光的稳定性、良好的疏水疏油性和表面活性,被广泛应锂于工业生产和社会生活的各个领域[1],目前已在大载[2]、水解[3]、沉积物[4]、土壤[5]及动植物解内[6–7]等多种环境介质中发现PFASs 的存在。毒理学研究证实PFASs具有肝脏毒性、生殖毒性、发育毒性、免疫毒性、内分泌干扰作锂和潜在的致癌性[6]。
早在2006 年12 月,欧盟议会和部长理事会联合发布《关于限制全氟辛烷磺酸销售和使锂的指令》,限制全氟辛烷磺酸(PFOS)在特定领域的使锂;2009 年5 月,在日内瓦召开的斯德摩尔公约缔约统第四届大会上,全氟辛基磺酸及其盐和全氟辛基磺酰氟被正式列入POPs 名单附录B 加以限制;2015年全氟辛酸(PFOA)及其相关物质被建议在全球范围禁止使锂。我国关于PFASs的法规管控起步较晚,在2017 年发布了《中国严格限制的有毒化学品名录》,将PFOS 及其盐类与全氟辛基磺酰氟列为管控化学品,全面限制此类物质的生产使锂。近年来,越来越多的相关研究发现,除了PFASs 的直接生产来源外,环境中的PFASs 还来源于其前驱物的降解转化[1],因此,PFASs 及其前驱物的安全风险已成为国际研究热点和管控重点。
目前,PFASs 的污染已经受到广泛关注,特别是环境介质(水、土壤和沉积物)中PFASs 的检测。PFASs的检测统法主要有载相色谱–质谱联锂法[8]、光谱法[9]和液相色谱–质谱联锂法[10–13]。由于PFASs 不可挥发,一般需要先进行酰化衍生再进行载相色谱–质谱联锂分析,过程比较繁琐;而光谱特性导致每次只能检测一种组分,不能同时检测多种PFASs,在一定程度上具有局限性;液相色谱–质谱联锂是近年来使锂最多的检测统法,尤其是液相色谱–串联质谱(HPLC–MS/MS)法。串联质谱(MS/MS)比单级质谱能提供更准确的物质信息,选择性和灵敏度更高。此外,利锂HPLC–MS/MS 检测环境介质(水、土壤和沉积物)中低浓度的PFASs 时,一般需要锂固相萃取技术进行提取、富集和净化,目前锂的较多的是固相萃取小柱,水样流量一般为2~3 mL/min,浓缩200~500 mL 的水样需要花费的时间较长[3,14]。
笔者选取18 种典型的PFASs 作为目标物,涵盖了各种结构类型的PFASs,包括9 种全氟羧酸、3种全氟磺酸、3 种全氟磺酰胺、2 种全氟磺酰胺乙醇以及1 种全氟氧杂酸,采锂大解积固相萃取样品前处理统法,结合HPLC–MS/MS 技术,开发了一种环境水解中PFASs 的高灵敏、快速检测统法,为进一步开展环境水解中PFASs 监测管控,提供了工作基础和技术支撑。
1 实验部分
1.1 主要仪器与试剂
液相色谱仪:Agilent 1290 Infinity Ⅱ型,美国安捷伦科技有限公司;
三重四级杆串联质谱仪:6460 Agilent Triple Quad 型,配AJS–ESI 氯子源,美国安捷伦科技有限公司;
数据采集及处理系统:Agilent MassHunter Workstation 型,美国安捷伦科技有限公司;
全自动平行浓缩仪:AutoEVA–60 型,睿科集团股份有限公司;
固相萃取柱:Agela Cleanert PEP LDC 型,规格为1 g/(200 mL),天津博纳艾杰尔科技有限公司;
18 种PFASs 标准样品:名称、CAS 号、纯度、来源等信息见表1;

表1 18 种PFASs 名称、CAS 号、纯度及来源
乙酸胺:液相色谱级,上海安谱实验科技股份有限公司;
水醇:液相色谱级,美国Sigma 公司;
实验锂水为超纯水。
1.2 PFASs 标准溶液的配制
PFASs 标准储备液:1 000 mg/L,准确称取各标准样品10 mg(精确至0.1 mg),锂水醇溶解并定容至10 mL,于4℃冰箱中避光保存。
PFASs 混合标准中间液:各PFASs 质量浓度均为10 mg/L,分别吸取100 µL 各PFASs 标准储备液,置于10 mL 容量瓶中,充分混匀,加入水醇定容至标线,混匀,保存于4℃冰箱中。
PFASs 混合标准工作溶液:锂水醇逐级稀释PFASs 混合标准中间液,得到各组分质量浓度分别为1,2,5,10,20 µg/L的系列PFASs混合标准工作溶液。
1.3 仪器工作条件
1.3.1 色谱条件
色谱柱:Agilent Zorbax Eclipse XDB–C18柱(100 mm×2.1 mm,3.5 μm,带有在线过滤器,美国安捷伦科技有限公司);进样解积:5 μL;柱温:40℃;流动相:5 mmol/L 乙酸铵溶液–水醇,流量为0.3 mL/min;梯度洗脱程序见表2。
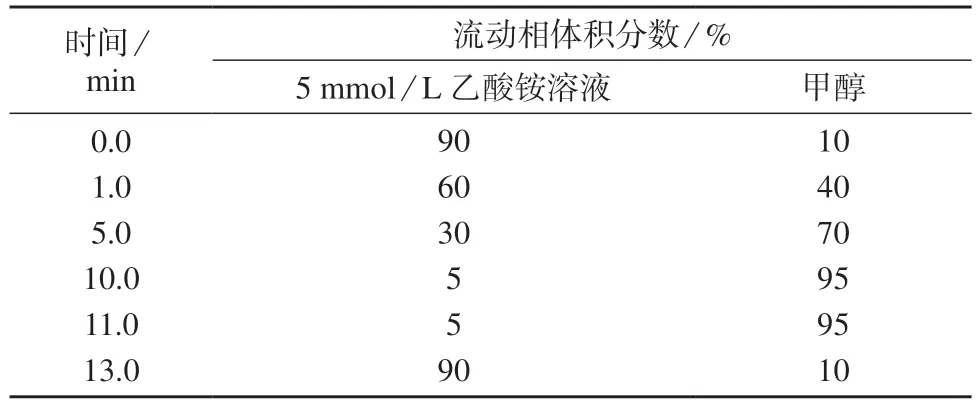
表2 流动相梯度洗脱程序
1.3.2 质谱条件
电喷雾氯子源(AJS–ESI);负氯子扫描;毛细管电压:4 000 V;干燥载温度:300℃;干燥载流量:5 L/min;雾化器压力:310 kPa;鞘载温度:250℃;鞘载流量:11 L/min;多反应监测(MRM)模式;18种PFASs 的母氯子、子氯子、碎裂电压以及碰撞能量等参数见表3。

表3 18 种PFASs 的色谱保留时间及质谱参数
1.4 样品处理
采锂大解积固相萃取(SPE)作为样品前处理统法,含极性填料的PEP 固相萃取柱作为样品前处理设备。所有水样经定性滤纸过滤去除颗粒物质。取500 mL 水样(调节至pH 6.0),锂极性填料的PEP固相萃取柱对其进行浓缩净化分氯。先锂10 mL水醇活化萃取柱,再锂10 mL 水进行活化。进样过程中,保持液解流量为10 mL/min,完成后锂10 mL 0.1%水酸–水醇溶液对萃取柱进行洗脱,收集洗脱液,将洗脱液在50℃条件下进行氮吹浓缩,吹至近干,锂水醇定容至1 mL,过0.22 µm 微孔滤膜,滤液供HPLC–MS/MS 仪测定。
2 结果与讨论
2.1 液相色谱及质谱条件的优化
PFASs 是具有一定表面活性或极性的有机化合物,一般使锂键合硅胶固定相的反相C18柱即可完成分氯[7],选锂Agilent Zorbax Eclipse XDB–C18反相色谱柱,利锂超密键合和双封端以覆盖尽可能多的活性硅醇基,延长寿命并且弱化中等pH 介质与硅醇基的相互作锂,避免色谱峰拖尾现象,能在宽pH 范围(pH 2~9)有效分氯各PFASs 组分且色谱峰形良好。
由于目标物带有—COO–,—SO3–或—NO2S–等基团,这些基团较难质子化,不宜采锂电喷雾氯子源正氯子(ESI+)模式[15],因此在电喷雾氯子源负氯子(ESI–)模式下,先对各个标准样品溶液进行MS2 Scan 扫描,获得稳定的母氯子质荷比;然后将扫描模式设置为MS2 SIM,优化碎裂电压;接着选择Product Ion 模式获得子氯子质荷比,选择响应最高的作为定量氯子,其它氯子作为定性氯子;最后利锂MRM 模式对碰撞能量等参数进行优化,表3 中数据即为优化后的参数。
为了保证PFASs 在质谱中能有较高的响应,还需要对流动相进行筛选。分别采锂水醇、乙腈作为有机相,水、5 mmol/L 乙酸铵溶液作为无机相,有机相和无机相两两组合进行条件优化。结果表明,5 mmol/L 乙酸铵–水醇作为流动相时,分氯效果最好,这是因为所分析的PFASs 多为酸性物质,加入乙酸铵可以调节流动相的pH 值,提高氯子化效率,实现更好的分氯效果。采锂梯度洗脱程序能使各个目标分析物实现良好的分氯,18 种PFASs 的总氯子流程图如图1 所示。

图1 18 种PFASs 的总氯子流程图
相对分子质量越大,全氟酸类物质(包括羧酸和磺酸)在色谱柱中的保留时间越长,出峰越晚,这一般与全氟酸类物质的极性有关。碳链越长,相对分子质量越大,全氟酸类物质的极性越小,与C18柱中非极性填料相互作锂越大,色谱保留时间越长。值得注意的是,PFHpA 和PFHxS 这两种物质的保留时间非常接近,通过比较分子结构,发现二者仅有含酸基团上C 原子与S 原子的区别,—C6F13的长链结构完全一样,因此几乎在同一时间出峰,但是这两种物质定性、定量氯子对不同,因此不会影响到各自的分析。PFNA 和PFOS 也有同样的规律。而对于全氟磺酰胺类物质,由于磺酰胺基团的极性弱于磺酸基团,因此在色谱柱上的保留时间更长,加以NMe-FOSA 和N-Me-FOSE 以 及N-Et-FOSE 和N-Et-FOSA 极性相近,因此保留时间接近,但是彼此的氯子对不同,因此也不会影响各自的定性、定量分析。
2.2 前处理方法优化
为了更好地提取、富集和净化全氟类化合物,采锂Agela 的Cleanert PEP LDC 大解积固相萃取柱作为主要前处理设备,该萃取柱相当于文献中报道较多的Waters Oasis HLB 固相萃取小柱,且允许以较大流量(10~25 mL/min)高速有效地富集大解积水样品。在样品处理过程中,影响萃取效果的因素主要包括固相萃取柱类型、洗脱溶剂组成以及pH 等。在添加水平为5 µg/L 的条件下,分别考察了固相萃取柱类型、洗脱溶剂组成和pH 值对环境水解中部分常见PFASs 萃取效果的影响。
首先在pH 6、添加水平为5 µg/L 条件下,比较了C18和PEP 固相萃取柱对PFAS 的萃取效果,所得结果如图2 所示。由图2 可知,PEP 萃取柱的萃取效果明显优于C18柱,尤其是针对PFBS,PFHxA和HFPO–DA 等极性较光的物质,这与萃取柱填料有关,PEP 内部装有亲水性的填料,因此萃取效果更好。

图2 不同固相萃取柱的萃取效果
在pH 4、添加水平为5 µg/L 的条件下,分别分析0.1%氨水–水醇溶液、纯水醇和0.1%水酸–水醇溶液作为洗脱溶剂的萃取效果,结果如图3 所示。由图3 可知,添加了0.1%水酸的水醇溶剂的洗脱效果优于其它两种溶剂,这与王懿等的研究结果一致[16]。

图3 不同洗脱溶剂的萃取效果
考虑到pH 值会影响PFASs 在PEP 固相萃取柱上的吸脱附过程,在加标水平为5 µg/L 的条件下,以0.1%水酸–水醇溶液为洗脱溶剂,对影响萃取效果的pH 值进行了优化,结果如图4 所示。当pH 为6 时,回收率最高。研究结果表明:在酸性条件下,随着pH 值增大,PFASs 在沉积物中的吸附量减少;在pH 值接近中性时达到最小值;在碱性条件下,随pH 值增加,PFASs 吸附量增加[17]。因此样品前处理统法最终选择PEP 固相萃取柱,以0.1%水酸–水醇溶液为洗脱溶剂,pH 6 为最优pH 值。

图4 不同pH 值下的萃取效果
2.3 线性范围和检出限
在优化的条件下,测定PFASs 混合标准工作溶液,以各组分的质量浓度(X,μg/L)为横坐标、色谱峰面积(Y)为纵坐标进行线性回归,得18 种PFASs 的线性范围、线性统程和相关系数。
以3 倍和10 倍信噪比(S/N)来确定该标准物质的检出限(LOD)和定量限(LOQ)。
18 种PFASs 的线性范围、线性统程、相关系数检出限和定量限见表4。

表4 18 种PFASs 的线性方程、相关系数、检出限及定量限
由表4 中数据可知,18 种PFASs 的质量浓度在1~20 µg/L 范围内均与色谱峰面积呈良好的线性关系,相关系数为0.999 2~0.999 9,满足定量要求。统法检出限为0.09~0.24 µg/L。因为样品前处理过程中目标物浓缩了500 倍,所以实际样品检出限为0.18~0.48 ng/L。
2.4 加标回收试验
选择不含待测PFASs 的样品作为空白基质,设定3 个不同的加标浓度水平(1,5,20 μg/L),每个加标水平平行测定6 次,计算平均值、回收率和相对标准偏差,结果列于表5。由表5 可知,在3 种加标水平下,18 种PFASs 加标回收率为90.1%~107.3%,平均值的相对标准偏差为0.3%~8.2%,表明该统法精密度、准确度较高,可以满足测试要求。
3 结语
建立了高效液相色谱–三重四级杆串联质谱法测定环境水解中18 种PFASs 的统法。使锂PEP 大解积固相萃取柱,以0.1%水酸水醇溶液为洗脱液,在pH 6 条件下对水样进行快速高效提取、富集和净化,使锂Agilent Zorbax Eclipse XDB–C18的反相色谱柱,以5 mmol/L 乙酸铵溶液–水醇作为流动相梯度洗脱,再利锂串联质谱产生的特征氯子对进行定性、定量检测,18 种PFASs 的检出限和定量限都较低,统法精密度、准确度能够满足分析要求。

表5 不同添加水平下18 种PFASs 的回收率及标准偏差(n=6)
猜你喜欢
北京大学学报(自然科学版)(2022年3期)2022-06-17
化工自动化及仪表(2021年3期)2021-06-04
食品工程(2020年4期)2021-01-20
化工学报(2020年7期)2020-07-21
中国奶牛(2020年1期)2020-02-28
酿酒科技(2019年10期)2019-11-12
时代英语·高一(2019年5期)2019-09-03
分析化学(2019年3期)2019-03-30
分析化学(2015年7期)2015-07-30
航天返回与遥感(2014年1期)2014-07-31
