
南沙海区深海沉积物中细菌多样性分析
2020-03-12 02:15明红霞陈泉睿张春鑫关道明樊景凤
生物学杂志 2020年1期
苏 洁,明红霞,陈泉睿,张春鑫,关道明,樊景凤
(1. 国家海洋环境监测中心,大连 116023; 2. 国家海洋局近岸海域生态环境重点实验室,大连 116023; 3. 大连海洋大学 水产与生命学院,大连 116023)
深海是全球最大的独立系统,地球表面有超过50%的面积被深海大洋所覆盖[1]。深海沉积物是集化学物质和微生物于一体的特殊生态环境,同时海底沉积物还是营养元素进行生物地球化学循环的主要场所[2]。在深海沉积物中存在着活跃的微生物群落,负责介导有机碳的降解和转化,在海洋和全球碳的生物地球化学循环中起着重要的作用[3]。深海沉积物中的微生物总量在全球生物量的比重已超过10%[4]。研究深海微生物多样性不仅有利于开发利用新型微生物资源,也将为人类探索生命起源提供重要线索。随着大洋钻探计划(ODP)多个航次的进行,太平洋边缘海域沉积物中的细菌群落结构及其优势类群已被逐步发现[5-8]。然而,由于深海独特的环境特征如低温、高压、缺氧等,导致海底99%以上的微生物无法利用现有技术得到分离培养[9],这使得基于16S rDNA的分子生物技术成为调查海底沉积物中微生物多样性的一种重要手段。
目前,研究学者对南沙海区表层沉积物中细菌和古菌多样性有了一些初步的了解,但研究对象主要为可培养细菌[10-11]。鉴于南沙海区广阔的水域及其多变的生境,加之以往的研究还不能全面地反映南沙海区深海沉积物中细菌资源多样性,为探讨南沙海区深海沉积物中细菌多样性,本研究运用16S rDNA克隆文库的方法展开对南沙海区深海沉积物中细菌群落结构的研究。
1 材料与方法
1.1 材料
2013年5月采集位于南沙群岛东南海域的深海表层沉积物样品(海水沉积物界面以下约5 cm)。采用箱式采泥器在南海3个不同站位分别采集样品,样品具体的信息描述如表1。

表1 南沙沉积物采样站点及样品描述
1.2 方法
1.2.1 沉积物细菌总DNA的提取与纯化
沉积物细菌总DNA的提取参照本课题组前期建立的方法[12]。使用QIAquikPCR纯化试剂盒对DNA进行纯化。
1.2.2 细菌16S rDNA的PCR扩增
采用细菌16S rDNA的通用引物27 F和1492 R,反应条件参照前期研究[12]。扩增产物用1.0%(W/V)的琼脂糖凝胶电泳检测。
1.2.3 细菌16S rDNA克隆文库的构建及阳性克隆的筛选
采用蓝白斑筛选方法筛选阳性克隆子并构建16S rDNA克隆文库[13]。
1.2.4 DNA序列系统进化分析
应用MEGA5.0软件,采用邻位相接法(Neighbour-Joining)构建系统发育进化树。
1.2.5 16S rDNA克隆文库多样性分析
采用克隆文库的覆盖度(Coverage)、Shannon-Wiener 指数及Simpson指数3个指标对构建的细菌16S rDNA克隆文库进行多样性分析。
2 结果与分析
2.1 16S rDNA克隆文库测序
对从3个深海站位沉积物样品构建的16S rDNA克隆文库中共获得的565个阳性克隆子,其中NSA站位为204个阳性克隆子,NSE站位为161个阳性克隆子,NSI站位为200个阳性克隆子。对上述3个站位获得的阳性克隆子分别进行序列测定,将序列相同的克隆子定义为一个OTU,NSA、NSE和NSI 3个站位的OTU数目分别为25、18和22。
2.2 16S rDNA克隆文库细菌多样性指数
本实验采集的3个样品均来自深海,受陆源微生物的影响较小,因而可以较准确地反映出南沙海区微生物群落的多样性。南沙海区沉积物样品中细菌16S rDNA克隆文库的覆盖度都大于80%,部分接近90%,表明克隆文库中所包含的细菌种类占沉积物样品中全部细菌的比例已很高,库容已经足够(表2)。细菌16S rDNA文库的Shannon-Wiener指数值从高到低依次为NSI、NSA和NSE,说明3个站位中细菌多样性程度最高的NSI站位,其次是NSA站位,较低的为NSE站位。

表2 16S rDNA克隆文库细菌多样性指数
2.3 16S rDNA克隆文库系统发育分析
对南海NSA站位沉积物中测序得到的25个OTU用MEGA软件进行系统发育分析,结果表明细菌克隆文库中的大多数序列分属于24个类群,9个门(图1)。而群落优势菌依次为变形菌、浮霉菌和放线菌。其中,南海NSA站位共获得55个变形菌序列(27%),浮霉菌序列共获得32个(15.6%),放线菌序列共有28个(13.7%)。通过与GenBank文库的比对发现,细菌16S rDNA克隆子的分布中变形菌门里α-Proteobacteria共有16条同源序列,3条序列是来自培养菌株,同源性都较高(89%~96%序列相似性)。分别是从盐田中分离的玫瑰弧菌属(Rhodovibrio)细菌、从土壤中分离的生丝微菌属(Hyphomicrobium)细菌以及来自纯培养的叶瘤杆菌科细菌Parvibaculumlavamentivorans,其余全部来自各种海洋环境中未培养的克隆子。γ-Proteobacteria有18条同源序列,其中有1条是来自土壤中分离的喜热噬甲基菌属(Methylocaldum)细菌,同源性一般(86%序列相似性),其余17条序列全部来自各种海洋环境中未培养的克隆子。δ-Proteobacteria共有21条同源序列。其中2条同源序列来自可培养的菌株,其余的19条同源序列来自各种海洋环境中未培养的克隆子。放线菌类群共有28条同源序列,其中1条同源序列来自土壤中分离的链霉菌科细菌Streptacidiphilusthailandensis,同源性较低(82%序列相似性),其余26条序列与来自卡斯卡底边缘和Forearc海盆产甲烷水合物的深海沉积物中未培养的克隆子具有较高同源性(93%~99%序列相似性)。
NSE站位沉积物样品中细菌克隆文库中的大多数序列分属于16个类群,8个门(图2)。其中放线菌、变形菌和浮霉状菌为优势菌。放线菌序列共获得38个(23.6%),变形细菌序列共获得34个(20.4%),浮霉菌序列共获得27个(16.7%)。通过与GenBank文库的序列比对发现,放线菌类群共有38条同源序列,其中有26条序列与来自卡斯卡底边缘和Forearc海盆产甲烷水合物的深海沉积物中未培养的克隆子有较高的同源性 (91%~99%序列相似性)。其余全部来自各种海洋环境中未培养的克隆子。变形菌门中α-Proteobacteria共有10条同源序列,分别是从盐湖中分离的硝化单胞菌属(Nitrosomonas)细菌、从土壤中分离的根瘤菌属(Rhizobium)细菌。γ-Proteobacteria有15条同源序列,其中有6条是来自土壤中分离的草螺菌属(Herbaspirillum)细菌,同源性较高(93%序列相似性)。δ-Proteobacteria共有9条同源序列,其中5条同源序列来自可培养的菌株,其余的4条同源序列来自各种海洋环境中未培养的克隆子。浮霉菌类群共有27条同源序列,全部来自各种海洋环境中未培养的克隆子,其中15条同源序列分别来自日本海槽和地中海Kazan泥火山的深海冷泉沉积物,同源性较高(95%序列相似性)。

Phylogenetic tree and evolutionary distances are given by the neighbor-joining method. Numbers at each branch point indicate the percentage supported by bootstrap values based on 1000 replications. Bootstrap values>50% are shown at branching points. The same below
图1NSA站位沉积物中细菌的16SrDNA基因序列系统进化分析
Figure 1 Phylogenetic analysis of the bacteria derived from sediments of NSA site according to 16S rDNA gene sequence
NSI站位站位沉积物样品中细菌克隆文库中的大多数序列分属于21个类群,8个门(图3)。其中变形菌、放线菌和浮霉状菌为优势菌。变形细菌序列共获得45个(22.5%),放线菌序列共获得36个(18%),浮霉菌序列共有31个(15.5%)。通过与GenBank文库的序列比对发现,变形菌门中α-Proteobacteria共有16条同源序列,全部来自各种海洋环境中未培养的克隆子。γ-Proteobacteria有21条同源序列,其中13条同源序列来自从海底淤泥中分离的脱硫杆菌属细菌Desulfobacteriumindolicum,同源性较高(93%序列相似性),其余8条序列全部来自巴伦支海HaakonMosby泥火山和墨西哥湾高盐沉积物等,同源性较高(90%~97%序列相似性)。δ-Proteobacteria共有8条同源序列。其中4条同源序列来自可培养的菌株,其余的4条同源序列来自Forearc海盆产甲烷水合物的深海沉积物中未培养的克隆子。放线菌类群共有36条同源序列,其中有19条同源序列来自日本海槽和地中海Kazan泥火山的深海冷泉沉积物,同源性较高(96%序列相似性),有6条同源序列来自卡斯卡底边缘深海沉积物中未培养的克隆子。浮霉状菌群共有31条同源序列,全部来自各种海洋环境中未培养的克隆子,其中15条同源序列分别来自卡斯卡底边缘和Forearc海盆产甲烷水合物的深海沉积物中未培养的克隆子,同源性都极高(93%~96%序列相似性)。
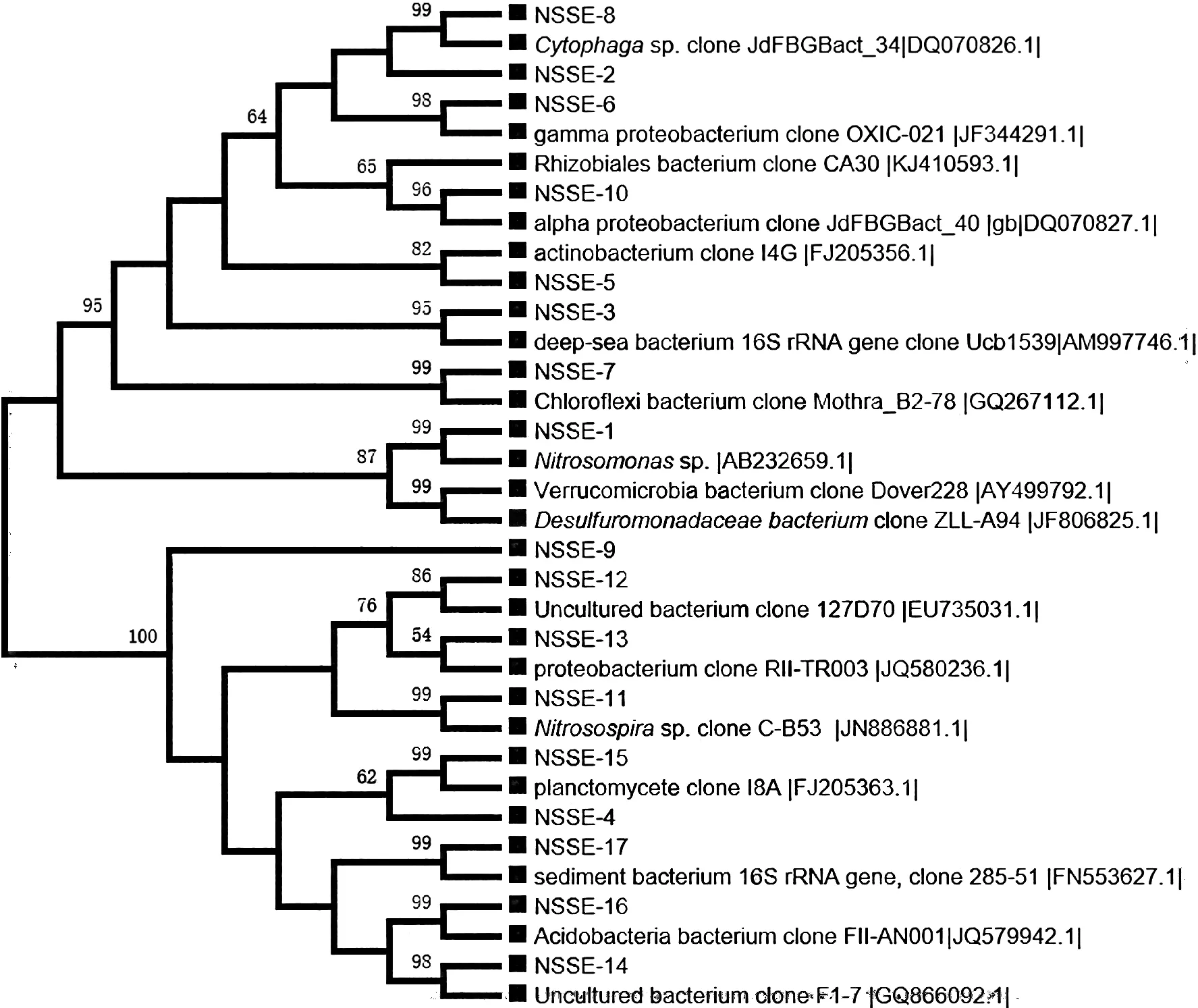
图2 NSE站位沉积物中细菌的16S rDNA基因序列系统进化分析
3 讨论
在本实验所选取的各站位深海沉积物中,广泛存在的主要优势细菌类群均包括变形菌类群、浮霉菌类群和放线菌类群。其中,本研究中构建的16S rDNA克隆文库涵盖了变形菌门中α-Proteobacteria(31.3%)、δ-Proteobacteria(40.3%)和γ-Proteobacteria (28.4%)3个亚门。可见,δ-Proteobacteria为优势亚群,这与南海西沙海槽沉积物中的结果一致[14]。然而在陆地土壤及海水环境中占优势的α-Proteobacteria在本研究中也占到了很高的比例,这与前期研究结果有较大差异[15]。但也有文献报道,α-Proteobacteria被发现为日本深海沉积物中的第二大优势类群[16]。深海环境中的优势类群γ-Proteobacteria,在本研究中也被证实为南沙海区深海沉积物中的第三大优势类群。此外,本文从3个深海站位沉积物样品中测序获得的16S rDNA序列中变形菌的优势亚群也有所不同,这些研究结果的差异主要是由于地理环境或气候环境不同所导致,而且即使在相近地点菌群也会有所差异。在本研究中,放线菌为另一优势类群,说明放线菌在深海沉积物中的丰度较高,这也与南海其他海区沉积物中的报道相一致[17-19]。除上述几种优势类群外,浮霉菌在本研究的细菌类群中也占到很大比例,这与孙慧敏等[1]在南海北部巴士海峡深海沉积物研究中的结果相一致。研究显示,该类群一般较多分布在浅海沉积物中,分析可能的原因为浅海来源的细菌由于物理迁移(风、潮汐等)等影响在深海海区沉积下来,并形成其优势菌。另外,我们从南沙海区获得的少数16S rDNA序列与发表在基因库中的16S rDNA序列表现出一定的差异,这一结果一方面说明细菌文库的多样性比较高;另一方面表明南沙海区仍存在一些未知种群。因此,研究中国南沙海区沉积物细菌的多样性,将进一步丰富对海洋细菌多样性的认识,为我们探究南海的细菌资源奠定基础。

图3 NSI站位沉积物中细菌的16S rDNA基因序列系统进化分析
猜你喜欢
猪业科学(2021年3期)2021-05-21
生态学报(2021年3期)2021-03-31
幽默大师(2020年10期)2020-11-10
房地产导刊(2020年8期)2020-09-11
家庭影院技术(2019年11期)2019-12-09
绿色科技(2019年14期)2019-11-19
中华诗词(2019年1期)2019-11-14
江苏农业科学(2019年5期)2019-09-02
猪业科学(2018年4期)2018-05-19
广东农业科学(2017年5期)2017-08-29
