
燃烧反应机理构建的双参数速率常数方法
2020-03-12 10:12李象远姚晓霞申屠江涛孙晓慧李娟琴刘明夏许诗敏
高等学校化学学报 2020年3期
李象远,姚晓霞,申屠江涛,孙晓慧,李娟琴,刘明夏,许诗敏
(四川大学化学工程学院,空天动力燃烧与冷却教育部工程研究中心,成都 610065)
燃烧反应机理是描述燃烧反应过程的基本方法. 基于燃烧反应机理的动力学模拟以及三维燃烧流场数值计算是湍流燃烧的基本研究手段. 由于燃烧反应苛刻的温度和压力条件,流场结构测量十分困难,燃烧数值模拟是燃烧流场全息化描述的重要手段.
19世纪末,化学反应动力学得到了迅猛发展,多位学者根据实验结果提出了化学反应速率常数(k)对温度(T)的依赖关系(表1)[1],其中,Arrhenius方程为
k=Aexp(-E/RT)
(1)

Table 1 Temperature-dependence equations of rate contants
式中:A为指前因子;T为温度;E为活化能;R为普适气体常数. 因其简单的形式、明确的物理意义和对实验结果的合理描述,成为化学反应动力学的基本公式. 后来,建立了简单碰撞理论和过渡态理论等理论模型来推演速率常数对温度的依赖关系. 按简单碰撞理论,双分子反应的速率常数表示为
k=AT1/2exp(-Ec/RT)
(2)
式中:Ec为反应碰撞阈能. 按过渡态理论,双分子反应速率常数表示为
k=ATexp(-H≠/RT)
(3)
式中:H≠为反应活化焓. 两种模型表明,Arrhenius方程的速率常数对温度的依赖关系是合理的. 但是,在较大的温度范围内,特别是在高温区域内,反应分子可能发生电子激发、非绝热跃迁等现象,Arrhenius方程的精确性则可能存在问题.
20世纪80年代,随着计算技术的进步和化学反应动力学模拟的需要,燃烧复杂反应的动力学机理得到了快速发展. 在反应机理构建之初,为了更好地拟合一些反应的速率常数对温度的依赖关系,Tsang等[2]在式(1)的指前项中引入T2~4项以得到更好的拟合结果,促使扩展的Arrhenius方程得到了广泛应用,即
k=ATnexp(-E/RT)
(4)
式中:n为温度指数;E仍然具有能量量纲,但不再具有活化能的物理意义.Tn项中T为纯数。对于一组已知的有限实验数据,用式(4)来代替式(1)可得到更高的拟合精度,因此在Tsang等[2]提出T2~4指前因子校正后,燃烧反应机理动力学速率常数的三参数形式被固化下来,成了复杂反应机理动力学速率常数的标准k~T形式,但n的取值范围不加限制,不再是简单碰撞理论和过渡态理论的1/2和1. 基于此动力学速率常数表达形式,人们发展了各种反应动力学模拟软件.
尽管式(4)比式(1)具有更高的拟合精度,但式(4)同时带来了一系列问题. 按式(1)描述,k对温度的依赖关系在整个温度范围内单调上升或下降,而式(4)实际上是在式(1)形式上引入一个“杠杆”因子Tn,n取值不同将导致速率常数对温度依赖的复杂化. 从式(4)可知,k~T曲线的斜率为
dk/dT=ATn-2(nT+E/R)exp(-E/RT)
(5)
式(4)将在T=-(E/nR)处取得极值(n≠0),n值非零将可能导致k~T曲线具有很大的曲率,这样用有限温度区间的数据训练出的三参数模型,应用于更高温度区间时失去了外推的依据,而式(1)由于具有直线形式:
lnk=lnA-(E/RT)
(6)
在一定的温度区间实现外推是可行的. 此外,式(4)所示的三参数形式可能导致同一套原始参数经不同研究者拟合出不同的结果,给基元反应动力学参数的比较带来困难,导致无法判断机理的优劣. 表2列出了国际上常用的燃烧核心机理的动力学参数. 可以看出,相同反应在不同机理中A,n,E参数相差很大. 从表2无法判断不同机理给出的速率常数的合理性,无法猜测速率常数对温度依赖的曲线形状,无法了解反应的能垒信息. 这样的参数化将造成机理构建的混乱. 事实上由于机理之间无法基于反应动力学进行比较,导致了燃烧反应机理的多样化和随意性,给机理的鉴别和统一造成困难. 由于活化能等物理量的缺失,机理整体优化只能是盲目试差,严重地影响了燃烧反应机理的发展[3].
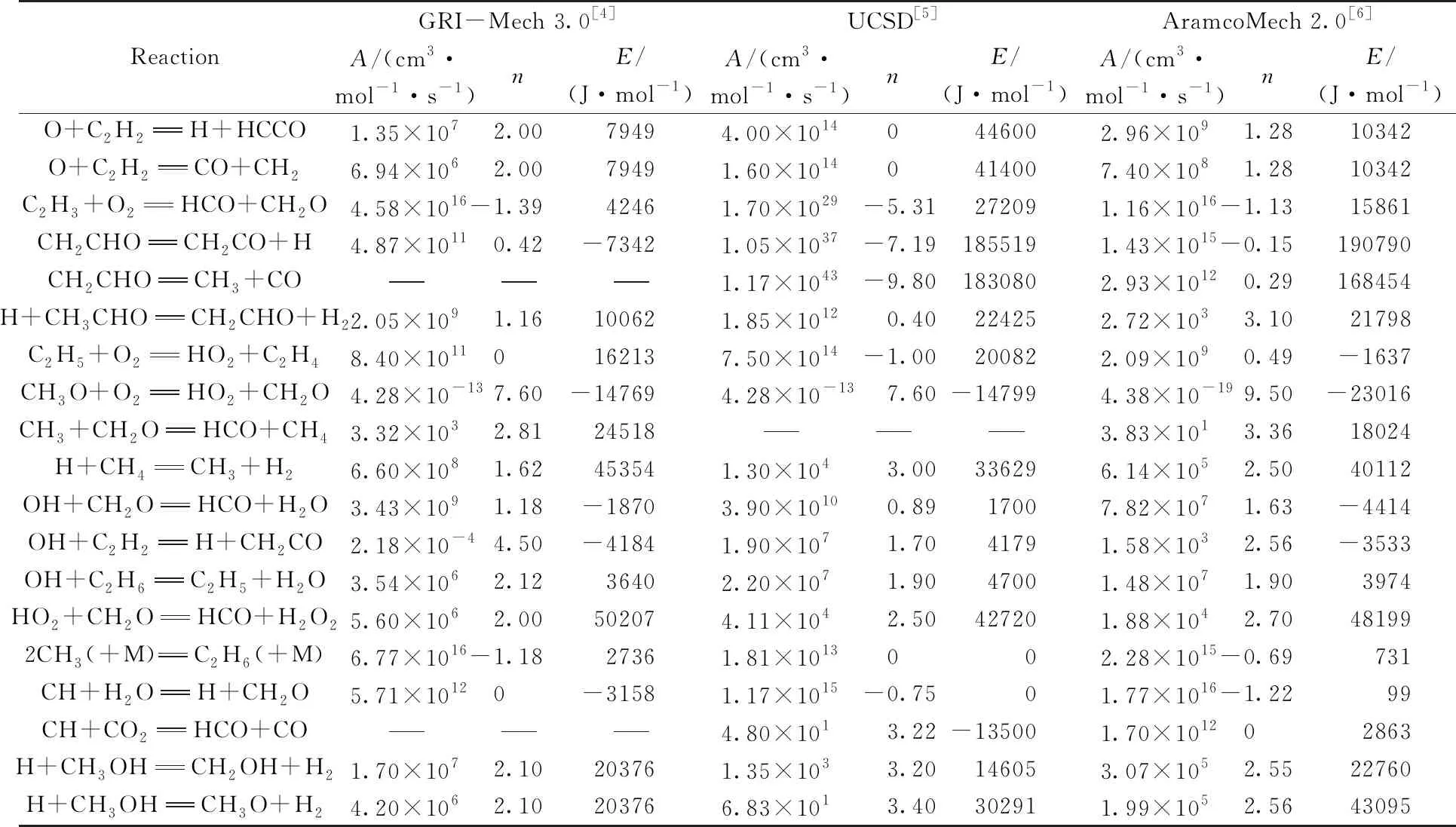
Table 2 Kinetic parameters in popular core mechanisms
由于动力学研究的困难和科学水平所限,不同研究者给出不同温度下的实验速率常数差别很大,且这些数据多属于中低温. 从动力学数据库如NIST[7]收集的基元反应实验数据判断,反应多采用Arrhenius方程描述,而在高温段的数据多采用化学反应速率的RRKM理论等方法计算得到. 与其它物理量不同,化学反应速率常数较难直接测量,通常要结合实验和动力学模型来获得. 由于高温高压等极端条件下测量的困难,精确的实验数据十分匮乏,因而对理论计算缺乏有效验证,有时只能通过动力学模拟获得高温点火延迟等宏观实验参数来判断机理的合理性.
1 实验方法
无论是实验测量还是理论计算,速率常数的精度仍然不高. 在有限的温度如低温范围内的基元反应,双参数的Arrhenius方程应该可以给出足够精确的描述. 如果对数据采用更为复杂的模型,由于训练样本的精度和温度范围所限,将会导致过拟合现象.

AramcoMech 2.0:k=1.14×1010T1.08exp(-2317/RT)
(7)
UCSD:k=1.66×1013exp(-3443/RT)
(8)
需要注意,指前因子中的T实际上是T/K,为无量纲纯数. 仅从两个表达式无法判断速率常数差别,但由lnk~1/T的线性关系图[图1(A)]可见,AramcoMech 2.0中的三参数结果在低温范围内lnk~1/T基本仍呈直线关系,三参数的过拟合导致高温区速率常数偏高,与UCSD机理的直线关系以及采用NIST数据库给出的动力学参数[7](均为Arrhenius型)取平均获得的直线关系预测的结果存在较大的偏差.
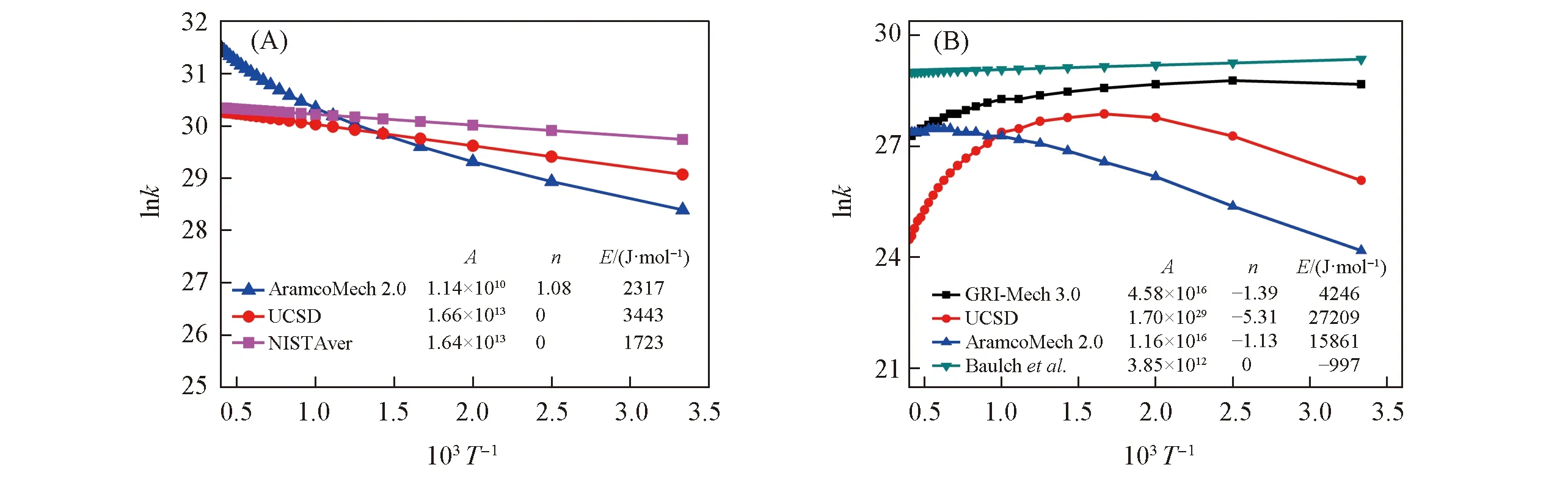
Fig.1 Temperature dependence of reaction rate constants for HO2+HH2+O2(A) and C2H3+O2CHO+CH2O(B) by three-parameter fitting and two-parameter fitting
504~923 K:k=4.3×1013exp(-43639/RT)
(9)
(10)
本文用文献原始数据做线性拟合结果稍有差别,分别为
504~923 K:k=1.7×1014exp(-56781/RT)
(11)
880~2495 K:k=1.9×1014exp(-60822/RT)
(12)
Sutherland等[19]发现两条直线不完全重合,便将速率常数在全温度范围内拟合成三参数形式:
504~2495 K:k=5.1×104T2.67exp(-26317/RT)
(13)
此速率常数表达式几乎在所有的机理中被采用. 但是,由于Tn的引入,26317 J/mol已经不再具有反应活化能垒的意义. 本文对图2离散速率常数做线性拟合,得到的双参数速率常数为
504~2495 K:k=2.6×1014exp(-60584/RT)
(14)
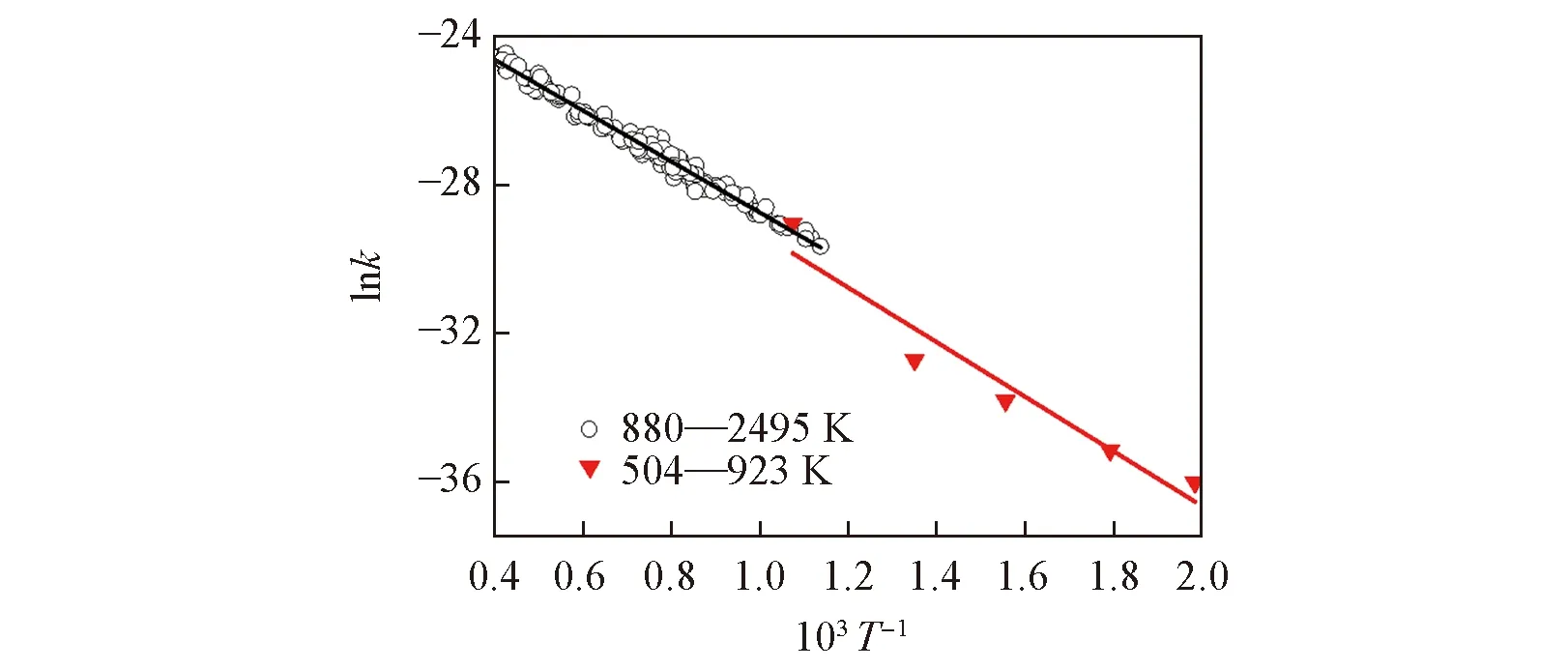
Fig.2 Flash photolysis-resonance fluorescence(504—923 K) and flash photolysis-shock tube(880—2495 K) rate constants of O(3P)+H2OH+H
相关系数R2=0.98338,并未降低拟合精度,但保留了活化能的可比性[9].
由此可见,燃烧反应机理速率常数的三参数表达式存在过拟合问题. 与单个反应的参数拟合不同,在复杂的多步反应机理中采用过拟合参数形式,将导致不同研究者发展的机理无法比较,机理整体优化变量空间大幅度增加,复杂的三参数曲线外推缺乏依据等问题.
1.1 线性拟合方法
通常,燃烧反应机理均需进行整体优化,即对标实验参数如点火延迟、火焰传播速度、燃烧过程的物种浓度演变等进行整体优化. 如果采用式(6)的Arrhenius双参数体系直线形式,在多步反应复杂机理的整体优化中,可在实验误差允许范围内,通过A来调整直线的截距,通过调整活化能E来调整直线斜率. 对大多数反应,A与化学键振动频率等物理量相关,对反应类型并不敏感,很多双分子反应的A取值在1013~1015cm3·mol-1·s-1范围内,大大压缩了整体机理优化的变量空间,使机理优化的参数调节主要为活化能的调整. 对于一般基元反应,可以根据反应能垒的理论计算值等来估计调整范围. 相比之下,由于三参数形式速率常数的A,n,E没有物理意义,对M个反应,参数变量约为3M个,对这些参数的调整实际上是盲目试差,目前采用三参数的复杂机理的整体优化通常只对A进行调节.
基于式(6)的线性拟合结果具有唯一性,目前的线性拟合多采用最小二乘法. 非线性拟合相对复杂,为了实现非线性数据拟合,首先要定义函数,在实际问题中,通常要根据离散点作图来寻找曲线类型,并给出拟合参数的初始值. 实际上,如果反应速率常数为非Arrhenius型,首先需要确定速率常数温度依赖关系的曲线形式,而式(4)所示的三参数曲线和真实曲线形式不一定相同. 另外,如果采用非线性拟合,优化结果受参数初值影响. 由于A,n,E不再具有物理意义,化学动力学基于反应能垒的理论体系将变得没有意义.
作者是科学研究的主体,通过对发文作者及其合作网络的结构特征进行分析,可以反映该领域研究的核心作者群及合作关系。本文将获取的文献数据导入CiteSpace V中,选择网络节点(node types)为Author,以关键路径(pathfinder)算法,数据抽取对象为Top50,设置时间区间(time slicing)的值为1,生成了作者共引聚类知识图谱(如图3)。
1.2 单分子反应的处理方法
燃料燃烧包含燃料裂解和氧化两个步骤. 裂解反应通常为单分子反应. 为了解释单分子反应速率常数的压力相关问题,Lindemann等[10]提出单分子反应机理,即

(15)
选取A*为准稳态物种,利用稳态近似可以得到:

(16)
在高压时

(17)
反应体现为一级.k∞=k1k2/k-1. 在低压时
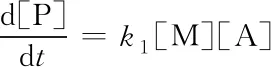
(18)
反应体现为二级. 将式(16)改写为d[P]/dt=k[A],有效速率常数k可表示为
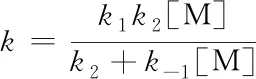
(19)
或
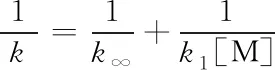
(20)
式(20)表明单分子反应速率常数的倒数1/k与1/[M]成正比. 由于压力和浓度的关系,发现在压力过渡区即所谓Fall off区,式(20)对单分子速率常数的预测存在偏差,进而提出了各种校正.
Troe因子校正方法是对式(20)乘上一个校正因子,即[11]
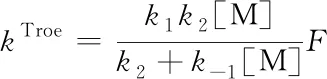
(21)
F是一个经验参数,由下式确定:

(22)

(23)
其中
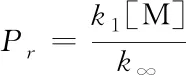
(24)
校正引入了经验参数:c,n,d,α,T***,T**,T*. 对于高温条件下的自由基反应,由于缺乏足够的样本数来训练校正模型,这些参数的引入存在随意性.
与Troe参数的校正公式不同,Plog方法[11]是一种描述反应速率的压力依赖性的通用方法,该方法是基于在给定两个压力下的反应速率取对数后直接插值,依据对数插值确定的速率常数,用扩展Arrhenius方程的三参数速率常数来描述不同压力下的速率. 该方法在介于给定压力点pi和pi+1间的压力p的速率常数k用插值表达式表示:
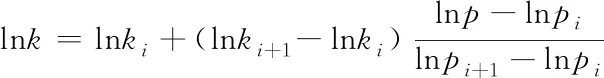
(25)
式中:ki和ki+1分别为pi和pi+1压力下的速率常数. Plog多项式提供了一种计算介于pi和pi+1间任意压力下的速率常数k的方法. 对多势阱多通道反应,表观速率常数与压力的关系较复杂,Troe校正公式难以应用,而对数内插的Plog在此时使用不受限,相比而言,Plog多项式求得的压力相关速率常数通常较Troe参数的校正公式使用范围更广,若气体混合物的成分不变,给定的压力点足够紧密时,Plog多项式也会得到更加精确的速率参数[12]. 但是,Plog方法求得的压力依赖速率常数的准确性需要足够多不同压力的速率常数值来保证. 在很多反应速率常数精度不高的情况下,个别反应动力学参数的精确表示难以使机理的预测精度获得明显改善.
在高精度电子结构计算的基础上,结合RRKM理论[13]和主方程方法[14]是获得压力相关速率常数的一种方法. RRKM理论是微正则过渡态理论,其微正则速率常数表达式为

(26)

(27)
式中:χ(E,t)是反应物与三体M发生碰撞后,在时间t时刻的能量分布,具体可以用一维主方程表示,即

(28)
目前,在燃烧反应机理中关于压力相关速率常数的计算,主要还是以一维的主方程为主. 一方面,Miller 等[15,16]研究发现,很多情形下一维与二维的结果基本一致. 另一方面,一维主方程比二维主方程更加简化.
林德曼机理是对单分子反应压力相关速率常数的一种模型近似,能用于实验验证的速率常数有限且多为低温数据,不同反应速率常数差别较大,无论是Plog列表还是对林德曼机理的Troe参数校正都是建立在一个较为复杂的经验公式或参数化拟合基础上,但用于训练这些校正模型的实验数据却非常有限. 对于大量单分子反应,无论实验还是理论计算,这种校正可能已经进入实验误差范围,如Troe参数化方案通常情况下F的值在0.2~1之间,对改善复杂反应速率常数精度意义不大.
无论是速率常数的三参数拟合还是对林德曼机理进行压力相关速率常数校正,实际上都是试图用一个更精确更复杂的模型来描述实验精度有限的速率常数,但由于燃烧机理是一系列反应的集合,且由于化学动力学和热力学参数的精度所限,一些“基元”步骤的动力学参数数据甚至是基于估计或按反应类型给出,要保证动力学模拟结果与宏观实验测量结果相吻合,还需要对机理进行整体优化,而个别反应速率常数的高精度不具有实际意义.
基于上述理由,本文构建的机理体系不引入Troe因子校正和Plog列表,而只给出高、低压极限速率常数k∞和k1,并根据式(19)计算压力相关速率常数k.
1.3 双参数核心机理
将较为成熟的UCSD机理中所有反应的动力学参数在宽温度范围区间内通过式(6)简单拟合成Arrhenius双参数形式,而不考虑反应的原始温度范围,得到机理双参数化的UCSD-R.
此外,将UCSD机理中的全部C0反应替换成本课题组[17]提出的基于极小化网络方法构建的双参数H2机理. 并将其它C1以上反应的参数重新处理,对非Arrhenius双参数形式的速率常数,根据原始文献,将动力学参数拟合成Arrhenius双参数形式. 一些非基元步骤缺乏实验参数,这类反应的速率常数具有较大的任意性,采用UCSD机理中三参数形式的速率常数值,通过式(6)线性拟合获得速率常数的双参数形式. 基于以上方法获得了双参数化初步核心机理UCSD-SC. 基于UCSD-R和UCSD-SC机理,进行了动力学模拟验证.
2 结果与讨论
点火延迟时间用来衡量燃料实际燃烧特性,同时也是验证燃烧反应机理合理性和准确性的重要指标. 实验可以通过激波管[18]测量燃料的点火延迟时间. 采用UCSD-SC机理、UCSD-R机理以及原始UCSD机理,用CHEMKIN-PRO程序包[19]模拟Petersen等[20]的甲烷燃料激波管点火延迟实验,结果见图3. 由图3(A)~(C)可见,简单双参数形式的UCSD-R机理的模拟结果与原始三参数形式的UCSD机理接近. 这说明三参数速率常数并不能明显改善计算结果,但双参数机理能还原活化能物理意义,同时大幅缩小参数变量空间,为机理的整体优化提供了便利. 从图3还可以看出,UCSD-SC结果和UCSD-R结果不同,这是由于UCSD-SC和UCSD-R的C0机理不同.

Fig.3 Ignition delay time by UCSD,UCSD-R,UCSD-SC and the experimental data(A) 100% CH4,p=1.09 MPa; (B) 80%/20% CH4/H2,p=2.14 MPa; (C) 60%/40% CH4/H2,p=2.36 MPa.

Fig.4 Ignition delay time by UCSD,UCSD-R and the experimental data(A) 6.25%C2H4/18.75%O2/75%N2,p=0.6—0.83 MPa; (B) 6.25%C2H4/18.75%O2/75%N2,p=1.12—1.61 MPa; (C) 11.76%C2H4/17.65%O2/70.59%N2,p=0.64—0.82 MPa.

Fig.5 Ignition delay time by UCSD,UCSD-R and the experimental data(A) 30%C2H6/70%H2,p=0.12 MPa; (B) 30%C2H6/70%H2,p=0.41 MPa; (C) 30%C2H6/70%H2,p=1.62 MPa.
还用UCSD-R机理以及原始UCSD机理模拟了Penyazkov等[21]的乙烷以及Pan等[22]的乙烯燃料激波管点火延迟实验,结果分别如图4和图5所示. 可以看出,UCSD-R机理的模拟结果基本与原始三参数形式的UCSD机理接近. 但UCSD-SC机理对乙烷和乙烯等其它燃料的模拟预测还可能需要进一步优化. 需要说明的是,UCSD等流行的核心机理都是通过对标大量实验数据优化的结果,对UCSD做简单双参数化处理基本能够重复UCSD结果,若经进一步整体优化,本文提出机理的预测结果将得到进一步改善.
3 结 论
目前燃烧机理广泛采用扩展Arrhenius方程的三参数形式. 由于速率常数k对温度T依赖的三个参数,A,n,E,没有物理意义,导致燃烧复杂反应机理的动力学参数缺乏可比性,用低温条件获得的数量有限的k值拟合出的A,n,E,当n绝对值较大时,外推到高温区将可能导致大的偏差. 根据扩展Arrhenius方程进行三参数拟合的另一个问题是过拟合问题. 在复杂的燃烧连串反应机理中,反应速率常数来源于实验测量和理论计算,而速率常数一般不能通过实验直接测量而要依据相关动力学模型,速率常数的理论计算更要引入若干假设. 精确度有限的实验数据,在全温度范围采用三参数拟合,存在过拟合现象. 目前的燃烧机理构建,不加区别地统一采用扩展Arrhenius方程形式来拟合所有的k~T曲线,显然是不合适的. 机理的整体优化中,采用Arrhenius方程的双参数速率常数,由于A对反应不敏感,机理优化可聚焦在活化能的调整,变量空间大幅度压缩. 由于活化能有明确物理意义,且不同反应类型的活化能已经有大量数据可供参考,机理整体优化难度降低. 基于上述结论,本文提出在燃烧复杂反应机理构建中,速率常数采用Arrhenius方程的双参数形式. 作为比较,本文采用线性拟合方法对动力学原始数据进行处理,同时通过对Troe参数校正因子和Plog形式等压力相关反应处理方式的合理性和实际效果评估,采用简单林德曼机理来表达单分子反应的压力相关速率常数. 本文在国际流行的UCSD机理基础上,构建了UCSD-R和UCSD-SC机理,并模拟了Petersen等的甲烷、Penyazkov等的乙烷以及Pan等的乙烯燃料激波管点火延迟实验工况. 结果表明,UCSD-R机理得到的模拟结果与原始UCSD机理的模拟结果基本一致,而UCSD-SC机理在Petersen等人所做甲烷实验工况条件下的模拟结果也是合理的,但对其它燃料的模拟预测,机理还需要进一步优化. 以上结果说明了双参数速率常数的可靠性,为燃烧机理的参数比较和迁移提供了可能,同时为燃烧机理基元反应的动力学参数统一探索一条可行的途径.
感谢郭震宇博士对本文在单分子反应速率常数双参数化方面研究工作的贡献.
猜你喜欢
建材发展导向(2021年14期)2021-08-23
数学年刊A辑(中文版)(2021年1期)2021-06-09
国学(2020年1期)2020-06-29
中国煤层气(2019年2期)2019-08-27
中国医学影像学杂志(2018年9期)2018-10-17
摄影之友(影像视觉)(2017年10期)2017-11-07
摄影之友(影像视觉)(2017年1期)2017-07-18
环境与可持续发展(2017年2期)2017-04-06
新高考·高一物理(2016年3期)2016-05-18
云南中医学院学报(2014年5期)2014-07-31
