
Establishment and characterization of a new cell line derived from half-smooth tongue sole Cynoglossus semilaevis kidney*
2020-03-19 12:31LOUYananSUNBinZHANGLinaLIYongXIAOPeng
LOU Yanan , SUN Bin , ZHANG Lina LI Yong XIAO Peng
1 Key Laboratory of Experimental Marine Biology, Institute of Oceanology, Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao 266071, China
2 Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China
3 University of Chinese Academy of Sciences, Beijing 100049, China
4 Tianjin Weishan Road Middle School, Tianjin 300222, China
Abstract In this study, we established a new cell line ( Cynoglossus semilaevis kidney cells, CSK) from kidney of the half-smooth tongue sole ( Cynoglossus semilaevis). The cells were subcultured over 1 000 days and passaged for more than 100 times. Additionally, CSK cells were optimally maintained in Dulbecco’s modif ied Eagle medium nutrient mixture F-12 supplemented with HEPES, antibiotics, fetal bovine serum,2-mercaptoethanol, and basic f ibroblast growth factor. The optimum growth temperature for CSK cells was 25°C, and the cells showed a f ibroblast-like phenotype. Chromosome analysis revealed that CSK cells had a normal diploid karyotype with 2n=42. CSK cells were susceptible to Grouper nervous necrosis virus (NNV), and cytopathic eff ects were observed at 3-5 days postinfection. The NNV sensitivity of CSK cells was related to the high abundance of virions in the cytoplasm, as observed by electron microscopy.Additionally, CSK cells could be successfully transfected with a green f luorescent protein reporter plasmid,and f luorescent signals were easily observed. Finally, immunocytochemistry analysis showed that CSK cells were supporting cells. Overall, we established this new cell line, which may have potential applications in the identif ication of viral pathogens aff ecting the half-smooth tongue sole.
Keyword: kidney cell line; Cynoglossus semilaevis; viral multiplication
1 INTRODUCTION
In vitro cell culture has gradually developed into an indispensable tool in many f ields of biological research. Since the establishment of the f irst stable f ish cell line, a gonadal cell line (RTG-2 cells) from rainbow trout (Salmogairdneri) (Wolf and Quimby,1962), more than 280 other f ish cell lines have been established and used in many studies and applications.Cultivation of cells in vitro has promoted the advancement of knowledge in various f ields, including functional genomics (Crespo et al., 2013),carcinogenesis (Lakra et al., 2011), developmental biology (Webb et al., 2015), immunology (Clem et al., 1996), virology (Gamil et al., 2015), genotoxicity(Rocco et al., 2014), gene expression (Barman et al.,2014), diff erentiation, developmental potentiality(Chen et al., 2010), physiology (Rode et al., 1997),and transgenic applications (Fan and Collodi, 2002).From these cell lines, in vitro experiments have identif ied many f ish pathogens, thereby improving our understanding of f ish diseases.
The half-smooth tongue sole (Cynoglossus semilaevisGünther) is a commercially valuable f latf ish. Owing to its high nutritional content and appealing taste, tongue sole is one of the most important cultured f ish species in China (Liao et al.,2009). However, with increased scale of aquaculture for this species, disease has become a major limitation aff ecting the growth of the tongue sole culture industry. Recently, more research groups have started to focus on identif ication of tongue sole pathogens,and several pathogens, includingVibrioharveyi(Chen, 2012),Listonellaanguillarum(Zhang et al.,2009),V.rotiferianus(Chen et al., 2012),Photobacteriumdamselaesubsp. piscicida (Wang et al., 2007),Mycobacteriummarinum(Luo et al.,2018), nervous necrosis virus (NNV) (Li et al., 2014),and a novel virus (Xiao et al., 2015), have been reported to be associated with diseases in the tongue sole. Compared with diseases in other f ish species,those in the tongue sole exhibit more complicated epidemiologic features, making it diffi cult to prevent such diseases. A 4-year monitoring study showed that some bacterial strains could be isolated from diseased tongue sole, although no eff ective antibiotics have been developed to control diseases in the tongue sole(data not shown). Thus, additional studies are needed to improve our understanding of tongue sole diseases,and establishment of novel cell lines from the tongue sole may facilitate the identif ication and detection of viral pathogens in the tongue sole. Although several cell lines have already been established from the tongue sole, including a liver cell line (HTLC cells)(Ren et al., 2008), an embryonic cell line (CFEC cells) (Sha et al., 2010), a heart cell line (CSH cells)(Wang et al., 2010), a testicular cell line (CGTC cells)(Zhang et al., 2011), a head kidney cell line (TSHKC cells) (Zheng et al., 2012), and a gonad cell line(CSPMG cells) (Sun et al., 2015), none of these cell lines have been used for identifying viral pathogens.
Therefore, in this study, we established a cell line from the tongue sole and evaluated its characteristics,with the goal of using this cell line to study viral pathogens in this f ish species.
2 MATERIAL AND METHOD
2.1 Primary cell culture and subculture
Several healthy half-smooth tongue sole(C.semilaevis) weighing 200-250 g were obtained from New Continent Food Co., Ltd. The f ishes were maintained in laboratory aquaria for at least 2 weeks.The surface of the f ish was wiped with 75% v/v ethanol solution, and the kidney was removed aseptically, transferred to a dish, and washed with Dulbecco’s modif ied Eagle’s medium (DMEM)/F-12(DMEM/F-12 powder with 4.76 g HEPES, pH 7.2)containing antibiotics (400 U/mL penicillin, 400 μg/mL streptomycin). The kidney tissues were then washed three times with sterile phosphate-buff ered saline (PBS), minced thoroughly with scissors, and digested with 1-mL trypsin solution for 10 min.Subsequently, 2-mL DMEM/F-12 complete medium(DMEM/F-12 supplemented with 20% fetal bovine serum [FBS], 100 U/mL penicillin, 100 μg/mL streptomycin, and 10 ng/mL basic f ibroblast growth factor [bFGF]) was added to the dish. The mixture of cells and cell clumps was centrifuged at 2 200 r/min for 2 min, and the pellet was suspended in 1-mL DMEM/F-12 complete medium and transferred to T-25 tissue culture f lasks. The cells were incubated at 25°C in an incubator with a normal atmosphere. The next day, 1-mL DMEM/F-12 complete medium was added to the f lask. One half of the DMEM/F-12 complete medium was changed every 3 days.
When the cells around the tissue pieces formed a monolayer, the old medium was removed, and the cells were washed with PBS. Cell clusters were digested with 1-mL trypsin and cultured in the original culture f lask with 2-mL DMEM/F-12 complete medium. Dissociation was monitored under an inverted light microscope (Nikon TE2000-S) to ensure that cells had been released from the f lask surface. For 10 passages, the cells were cultured in the original culture f lask. After 10 passages, cells were subcultured and transferred to new T-25 f lasks at a split ratio of 1:2. The primary cells were initially maintained in DMEM/F-12 complete medium with 20% FBS.
2.2 Growth curve
For the growth study, the cells at passage 18 were seeded into 12-well plates at an initial density of 2×105cells/mL and incubated at 25°C for 8 days. One well of cells was digested each day with 0.25%trypsin, and the number of cells was counted using a hemocytometer. The experiment was performed in triplicate. For construction of cell growth curves, the cultivation time was set as the abscissa and the number of cells per milliliter was set as the ordinate.
2.3 Eff ects of temperature on cell proliferation
To analyze how temperature aff ected the growth of the cells, 2×104cells/mL at passage 22 were inoculated in three 12-well plates and incubated at 16°C, 25°C,or 30°C. Cell numbers were counted microscopically using a hemocytometer every day for 8 days. The experiment was performed in triplicate.
2.4 Chromosome analysis
For chromosome analysis, we used CSK cells at passage 23. Brief ly, the cells were seeded in T-25 f lasks and cultured at 25°C overnight. The cells were then treated with colchicine (1 μg/mL) for 3.5 h at 25°C. The old medium was removed, cells were trypsinized, and DMEM/F-12 complete medium was added to the f lasks. The cells were harvested by centrifugation (650×g, 3 min). The pellets were resuspended in 5-mL hypotonic solution (0.075 mol/L KCl) and incubated at 37°C for 30 min. The cells were then f ixed for 2 min in 1-mL precooled Carnoy’s f ixative (methanol:acetic acid=3:1), centrifuged at 1 000 r/min for 6 min, f ixed twice in 2-mL precooled Carnoy’s f ixative (15 min each time), centrifuged again, and resuspended in 0.5-mL precooled Carnoy’s f ixative. Finally, slides were prepared using the conventional drop splash technique (Freshney) and then air dried. Chromosomes were stained with 5%Giemsa for 30 min. One hundred photographed cells at metaphase were counted under a Nikon eclipse 80-I f luorescence microscope, and chromosome karyotype was analyzed according to the method of Levan et al.(1964).
2.5 Sex genotyping of CSK cells
A DNA extraction kit was used to extract DNA from the established half-smooth tongue sole kidney cell line. Brief ly, based on sex-specif ic sequences, we designed primers for the sequence characterized amplif ied region, as follows: sense primer,5′-CCTAAATGATGGATGTAGATTCTGTC-3′, and reverse primer, 5′-GATCCAGAGAAAATAAACCCAGG-3′ (Liu et al., 2014). In females, these primers yield two DNA fragments of 169 and 134 bp, whereas in males, these primers yield one DNA fragment of 169 bp. The polymerase chain reaction (PCR)conditions were as follows: initial denaturation at 95°C for 10 min; 35 cycles of 95°C for 30 s, 54°C for 30 s, and 72°C for 40 s; and termination of the reaction at 72°C for 10 min. The PCR products (8 μL) were resolved on 2% agarose gels with a Trans 2k plusⅡDNA marker.
2.6 Virus susceptibility
CSK cells were used to detect susceptibility to Grouper NNV. Brief ly, we seeded CSK cells at passage 26 into T-25 f lasks and incubated the cells overnight. When the cell conf luence was approximately 90%, the medium was removed, and the cells were washed twice with PBS. Next, 1-mL NNV solution diluted with DMEM/F-12 minimal medium was added to the f lasks. After 1 h, the virus solution was replaced with 3-mL DMEM/F-12 complete medium containing 5% FBS. The cytopathic eff ect (CPE) was observed every 12 h using an inverted microscope. For observation of the CPE, we collected NNV-infected cells, f ixed the cells with 2.5% glutaraldehyde in cacodylate buff er (0.1 mol/L,pH 7.4) for 4 h at 4°C, rinsed the cells three times with PBS (0.1 mol/L, pH 7.4), and postf ixed the cells with 1% osmium tetroxide in cacodylate buff er(0.1 mol/L, pH 7.4) for 2 h. The samples were then rinsed three times in PBS buff er for 10 min. We used graded ethyl alcohol (30%, 50%, 70%, 90%, and 100%) to dehydrate the samples and then embedded the samples in Epon8-12 epoxy resin. A diamond knife was used to cut ultrathin sections on a Reichert-Jung Ultracut-E microtome. Samples were then mounted on copper grids, stained with 2% uranyl acetate-lead citrate, and examined with a JEOL JEM-1200EX transmission electron microscope. NNVinfected cell supernatants were harvested and f ixed with 2.5% glutaraldehyde in PBS (0.1 mol/L, pH 7.4)for 15 min. Subsequently, the samples were mounted on copper grids, stained with 3% phosphotungstic acid, and washed with double distilled water three times. The copper grids were placed in a desiccator and then examined with a JEOL JEM-1200EX transmission electron microscope.
2.7 Cell transfection with the pEGFP-N1 reporter gene
CSK cells were seeded at a density of 1×106cells/well in 12-well plates and incubated at 25°C. When the cells reached 85%-90% conf luence, the cells were transfected with the pEGFP-N1 expression vector using Lipofectamine 2000 (Invitrogen, Carlsbad, CA,USA). Brief ly, 2-μL Lipofectamine 2000 was mixed with 98-μL Opti-MEM (Invitrogen) in a 0.5-mL sterile centrifuge tube. At the same time, 2-μL pEGFP-N1 (500 ng/μL) was mixed with 98-μL Opti-MEM (Invitrogen) in a 0.5-mL sterile centrifuge tube.After 5 min, the two solutions were mixed and then incubated together for 20 min. The cells were washed twice with PBS, and then the medium was substituted with Opti-MEM. The mixtures were then dropped into the wells, and cells were cultured at 25°C for5.5 h. The medium was then replaced with normal medium. After incubation for 24 h, the cells were washed twice with PBS, f ixed with 4%paraformaldehyde (PFA) for 10 min, and washed twice with PBS. The nuclei were then stained with 4,6-diamino-2-phenylindole (DAPI) for 15 min and washed with PBS twice. Finally, the green and blue f luorescence signals were observed under a f luorescence microscope (Zeiss LSM710).

Table 1 Antibodies used in the present study
2.8 Cryopreservation and recovery of cells
Cells at 85%-90% conf luence were used for cryopreservation. The cells were trypsinized and suspended in DMEM/F-12 complete medium and then centrifuged at 2 200 r/min for 2 min. The cell pellet was resuspended with the medium containing 10% dimethyl sulfoxide and 90% FBS. Cells were then dispensed into 2-mL sterile plastic tubes, kept in a NALGEBE Cryo 1°C Freezing Container(Invitrogen) at -80°C overnight, and transferred to liquid nitrogen (-196°C) the next day. When thawed,the cryopreserved cells were placed into double distilled water at 42°C until just thawed and then centrifuged at 2 200 r/min for 2 min. The cells were suspended in DMEM/F-12 complete medium,transferred to T-25 f lasks, and cultured at 25°C in a normal atmosphere incubator.
2.9 Immunocytochemistry
At passage 37, CSK cells were used for immunocytochemical analysis with specif ic antibodies for supporting cells. The cells were seeded at a density of 1×105cells/well in 12-well plates and cultured at 25°C. When the monolayer reached 75%-85% conf luence, the old medium was removed, and the cells were washed with precooled PBS twice and f ixed with 4% PFA. The cells were washed with PBS and perforated with 0.2% TritonX-100 for 15 min.The cells were then washed with PBS twice, blocked with 1% bovine serum albumin for 30 min, and incubated with primary antibodies (Table 1) at 4°C overnight. After incubation, the cells were washed with PBS twice and then incubated again with secondary antibodies (Alexa-Fluor 488-labeled Goat Anti-Rabbit IgG (H+L) and Alexa-Fluor 488-labeled Goat Anti-Mouse IgG (H+L)) in the dark for 1.5 h at room temperature. Subsequently, the cells were washed with PBS twice and stained with DAPI for 15 min. A f luorescence microscope (Zeiss LSM710)was then used to observe the results. For the negative control, the primary antibodies were omitted.
3 RESULT
3.1 Primary cell culture and subculture
Approximately 3-5 days after the tissue pieces and cell suspensions were inoculated onto the plates, the cells began to move out from the edges of the tissue blocks. When the cells grew to 90% conf luence, the f irst subculture was performed. After 10 passages, the cells were subcultured at a ratio of 1:2 for (3-7)-day intervals. The culture time for cells at early passages was longer than that for later cells. The cells showed a f ibroblast-like phenotype (Fig.1).
3.2 Growth curve
Cells were plated at a density of 2×105and incubated for 24 h. The growth curve of CSK cells is shown in Fig.2. After 4 days, the cell number reached 8.9×105cells/mL.
3.3 Eff ects of temperature on cell proliferation

Fig.1 Morphology characterization of the CSK cells

Fig.2 The CSK cells growth curve
At passage 22, CSK cells were able to grow at temperatures ranging from 16 to 30°C. The cell numbers were determined at diff erent temperatures for 8 consecutive days. The highest growth rate was obtained at 25°C, with cell numbers reaching 17.45×104cells/mL after 4 days, and the lowest growth rate was obtained at 16°C, with the cell number reaching only 12.17×104cells/mL (Fig.3). There were no obvious diff erences in the morphological characteristics of cells cultured under diff erent temperatures.
3.4 Chromosome analysis
Chromosome analysis of cells at passage 23 showed that there was a chromosome number of 42 for 43% of 100 metaphase cells (Fig.4c). During metaphase(Fig.4a), cells having a normal diploid number also exhibited a normal karyotype morphology (Fig.4b).
3.5 CSK cells being genetically male
The results revealed that the cells had a diploid karyotype consisting of 21 pairs of chromosomes; the female-specif ic W chromosome was absent (Fig.4a).Accordingly, we concluded that CSK cells were male cells. Furthermore, PCR analysis revealed that the CSK cells yielded only the 169-bp male DNA fragment, further conf irming that these cells had been derived from a male f ish (Fig.5).
3.6 Virus susceptibility
CSK cells were susceptible to NNV, and a CPE was observed at 3 days after NNV infection. The cells were shrunk and became circular, and the cell nucleus was black and dense (Fig.6).

Fig.3 Eff ects of temperature on the growth of the CSK cells
When we observed a typical CPE, the cells were collected for TEM analysis. We found scattered virus particles in the cytoplasm (Fig.7b-d) and changes in the cell morphology after virus infection, suggesting that the CSK cells were sensitive to NNV.
3.7 DNA transfection
The cells were successfully transfected with pEGFP-N1 using Lipofectamine 2000, and clear, strong green f luorescence signals were detected after 24 h (Fig.8).The effi ciency of transfection was about 26.9%,suggesting that CSK cells were suitable for transfection.
3.8 Cryopreservation and recovery of cells
CSK cells were cryopreserved at diff erent passages.After thawing and seeding into f lasks, the cells recovered with a survival rate of 70%-80%. The cells could grow to conf luence after 7 days.
3.9 Immunocytochemistry

Fig.4 Chromosome analysis of CSK cells at passage 23

Fig.5 Half-smooth tongue sole sex specif ic markers to identify CSK cells
Antibodies specif ic for epithelial cells and stromal cells were used for immunocytochemical analysis in CSK cells. RLT1572 detects endogenous epithelial cell adhesion molecule protein. RLT5053 and RLM3364 detect endogenous α-SMA protein. RLT1453 detects endogenous E-cadherin protein. RLT1275, RLM3095,and RLM3080 detect endogenous diff erent Cytokeratin proteins. The results showed that most cells presented f luorescence signal, which means CSK cells were mainly supporting cells (Fig.9).
4 DISCUSSION
With the expansion of aquaculture scale owing to the global demand for aquaculture products, the development of the aquaculture industry is facing increasing problems, particularly with regard to the frequency of new diseases. New sequencing technologies have detected novel viruses in diseased organisms. However, we have not obtained direct evidence demonstrating that these viruses are the cause of disease. Cell culture is an essential tool for biological research and has facilitated the identif ication of many viral pathogens from various hosts (John and Richards, 1999; Isshiki et al., 2004). Notably, in previous studies, many bacterial pathogens have been isolated from diseased tongue sole, although no eff ective antibiotics have been developed to control these diseases. Thus, we still have an incomplete understanding of tongue sole diseases, and additional cell lines are urgently needed to support further research in this f ish species.

Fig.6 NNV-infected cells
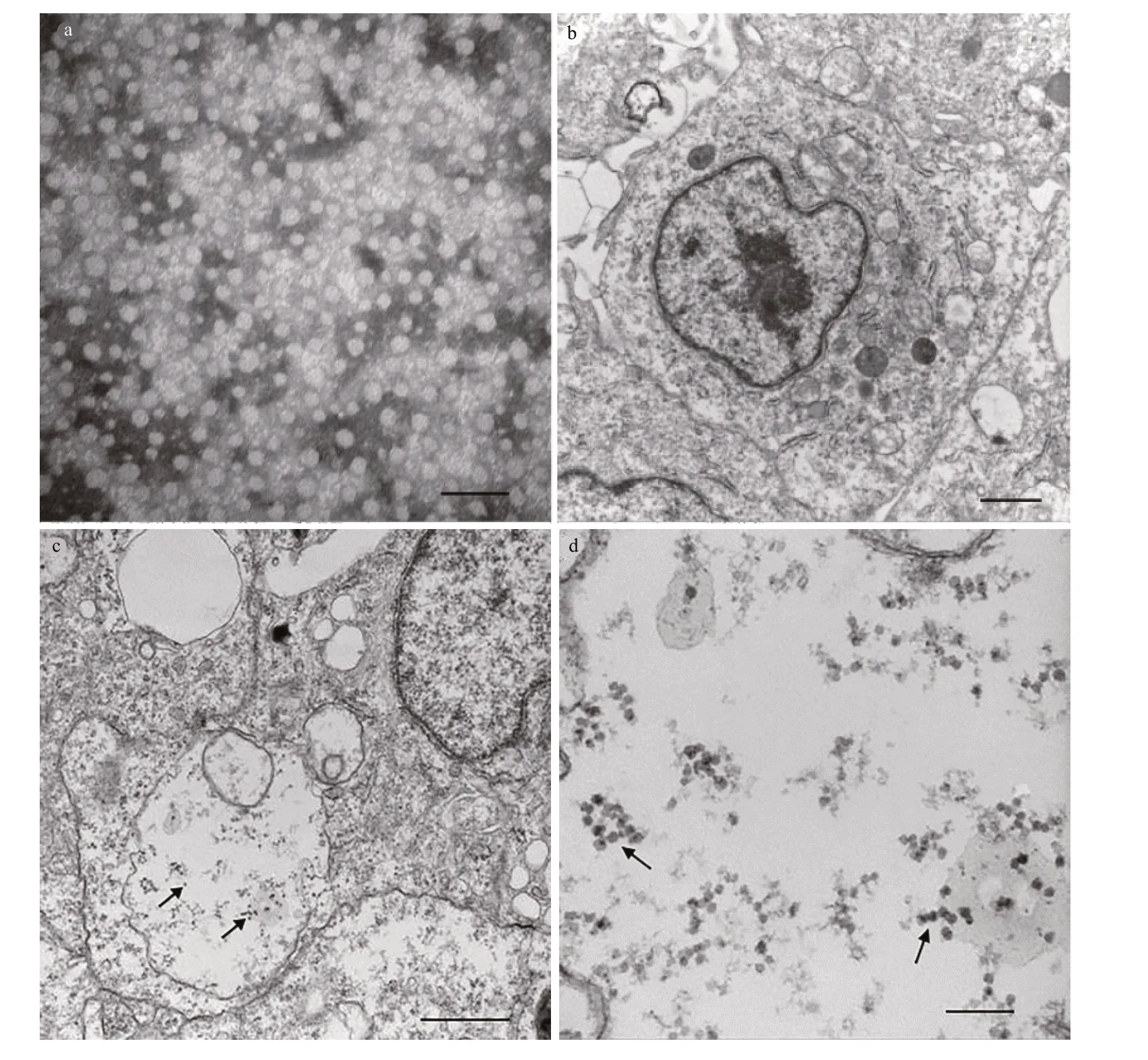
Fig.7 NNV infected CSK cell
In this study, a half-smooth tongue sole(C.semilaevis) kidney cell line was successfully developed and characterized, designated as CSK cells.We characterized the growth, transfection effi ciency,NNV susceptibility, and internal and external morphologies of the CSK cell line. Our results showed that CSK cells grew stably in DMEM/F-12 supplemented with FBS and bFGF. The cells were subcultured for 100 passages and showed a f ibroblasticlike morphology.

Fig.8 The expression of pEGFP-N1 in CSK cells

Fig.9 Immunocytochemical analysis of CSK cells by specif ic antibodies

Fig.9 Continued
CSK cells could grow under diff erent temperatures ranging from 16 to 30°C, with optimal growth at 25°C. Notably, this optimum temperature was the temperature at which the primary culture was established and was similar to that of other halfsmooth tongue sole cell lines (Ren et al., 2008; Sha et al., 2010; Wang et al., 2010; Zhang et al., 2011; Sun et al., 2015). These results also demonstrated that f ish cell culture is diff erent from mammalian cell culture,and f ish cells showed many advantages over mammalian cells (Bols and Lee, 1991). For example,f ish cells can be cultured at room temperature (16-30°C) and can be directly exposed to environment of the same atmospheric pressure. Additionally, f ish cells that adapt to a wide temperature range can be used for a wider range of applications, suggesting potential suitability for isolating both warm water and cold water f ish viruses (Nicholson et al., 1987).
From the well-spread metaphase chromosomes obtained in this study, we concluded thatC.semilaevishad a diploid chromosome number of 42 (Shao et al., 2010). Indeed, karyotype analysis revealed that 43% of the CSK cells possessed a diploid chromosome number of 2n=42, but lacked the heterogametic W chromosome. Furthermore, PCR analysis revealed that the CSK cell line did not have a female-specif ic marker, exhibiting the ZZ genetic constitution.
The susceptibility of CSK cells to NNV was evaluated by observation of the CPE. At 3 days after infection, cells inoculated with NNV exhibited morphological changes, including cell shrinkage and black and dense nuclei. Electron microscopy observations revealed that some virus particles were scattered throughout the cytoplasm of cells infected with NNV. As a viral pathogen to tongue sole (Li et al., 2014), NNV is also a causal agent of disease in many f ish species. Although some cell lines have been developed for reproduction of NNV (Chi et al.,2005), development of this cell line from tongue sole will provide more appropriate conditions for studies of viruses. The results showed that the CSK cell line was susceptible to NNV, providing better condition for virus research.
Kidneys are comprised of many specialized cells,which facilitate diverse activities. Renal cells are broadly classif ied as parenchymal (functional cells)or stromal cells (support cells), and these categories include diverse epithelial and mesenchymal cell types.The epithelial cells are arranged into working units called nephrons, and the mesenchymal cells occupy the intervening interstitial spaces between nephrons(Gerlach and Wingert, 2013). Our f indings demonstrated that CSK cells were mainly support cells.
In conclusion, we established a new cell line from the kidney of the half-smooth tongue sole. This cell line is expected to provide a useful tool for the identif ication of viral infections and for exogenous gene expression in the half-smooth tongue sole.Furthermore, this cell line may be instrumental in the analysis of mutant gene function in the context of kidney disease at a cellular level.
5 DATA AVAILABILITY STATEMENT
All data generated and/or analyzed during this study are available from the corresponding author on reasonable request.
 Journal of Oceanology and Limnology2020年2期
Journal of Oceanology and Limnology2020年2期
- Journal of Oceanology and Limnology的其它文章
- Contribution of surface wave-induced vertical mixing to heat content in global upper ocean*
- Upper ocean response to typhoon Kujira (2015) in the South China Sea by multiple means of observation*
- Inf luence of simulating deep-sea environmental factors on cathodic performance of seawater battery*
- Adsorption characteristics of chitooligosaccharides onto activated charcoal in aqueous solutions*
- Eff ects of hypoxia on survival, behavior, and metabolism of Zhikong scallop Chlamys farreri Jones et Preston 1904*
- Distinct inf luence of trimethylamine N-oxide and high hydrostatic pressure on community structure and culturable deep-sea bacteria*