
妥卡替尼的合成新工艺
2020-04-07 09:59张天军高军龙
山东化工 2020年5期
张天军,高军龙
(1.江苏恒瑞医药股份有限公司,江苏 连云港 222047;2.浙江先锋科技股份有限公司,浙江 台州 317021)
妥卡替尼(tucatinib),又称irbinitinib、ARRY-380或ONT-380,由Array生物制药公司自主研发,是一种人表皮生长因子受体酪氨酸激酶ErbB-2(也称为HER2)的口服抑制剂[1],正处于HER2+乳腺癌的II期临床研究[2],具有潜在的抗肿瘤活性。
目前该公司已报道过合成妥卡替尼的相关路线(如图1)[3],以2-氨基-5-硝基苄腈(2)为原料与N,N-二甲基甲酰胺二甲基缩醛(DMF-DMA)反应,经分离纯化后得到N'-(2-氰基-4-硝基苯基)-N,N-二甲基甲脒(3),收率87%。化合物3经催化氢化还原得到苯胺化合物4,收率90%。在-10℃下,化合物4与2-氨基-2-甲基-1-丙醇和1,1'-(硫代羰基)-二咪唑(TCDI)反应16 h,经柱色谱纯化后得到硫脲化合物5,收率为34%。化合物5与4-([1,2,4]三唑并[1,5-a]吡啶-7-氧基)-3-甲基苯胺(6)反应,再经柱色谱纯化后得到4-苯基胺喹唑啉化合物7,收率62%。化合物7经与NaOH和p-TsCl发生环合反应得到最终产物妥卡替尼(收率未报道)。该路线中间体的纯化大部分为柱层析方法,且由化合物4制备5这一步反应收率过低,目前只适用于药物化学阶段的实验研究,不适合妥卡替尼的放大制备。

图1 文献报道的妥卡替尼的合成路线
为了开发妥卡替尼更为实用的制备方法,本文设计了一种新的合成路线,如图2所示。分别制备了三种关键中间体,包括化合物6,喹唑啉-4,6-二胺化合物15和甲硫基-4,5-二氢恶唑化合物17。
化合物6的合成是基于对硝基苯化合物13[3-4]的优化而得。以2-甲基-4-硝基苯酚(8)和4-氯吡啶-2-胺(9)为起始原料,经重结晶纯化得到化合物4-(2-甲基-4-硝基苯氧基)吡啶-2-胺(10),收率64%。化合物10分别与DMF-DMA、盐酸羟胺反应,得到N-羟基-甲酰胺化合物12,产率为81%。再由三氟乙酸酐(TFAA)处理化合物12后得到三唑并[1,5-a]吡啶化合物13,其产率约为70%。化合物13经催化氢化还原得到化合物6,收率为93%,纯度为99.1%。
将2-氨基-5-硝基苄腈(2)与DMF-DMA反应,得到化合物合物3[3],收率88%。化合物6和化合物3在乙酸溶液中进行成环反应,得到化合物14,其产率为83%,纯度为99.3%。再将化合物14在室温条件下通过H2/Pd-C/THF的条件催化氢化还原制得化合物15,其收率为92%,纯度为99.5%。
甲硫基-4,5-二氢恶唑化合物17是基于文献报道经优化制备[5-6],以2-氨基-2-甲基-1-丙醇和1,1'-(硫代羰基)-二咪唑(TCDI)为原料,在CH2Cl2中经成环反应得到4,4-二甲基恶唑烷-2-硫酮(16),收率83%;化合物16与三氟甲磺酸甲酯反应,得到化合物17,收率81%。最后,将化合物17和化合物15溶于Cs2CO3/DMF体系,在125 ℃下反应20 h,经重结晶纯化得到最终目标化合物1,产率为76%,纯度为99.5%。
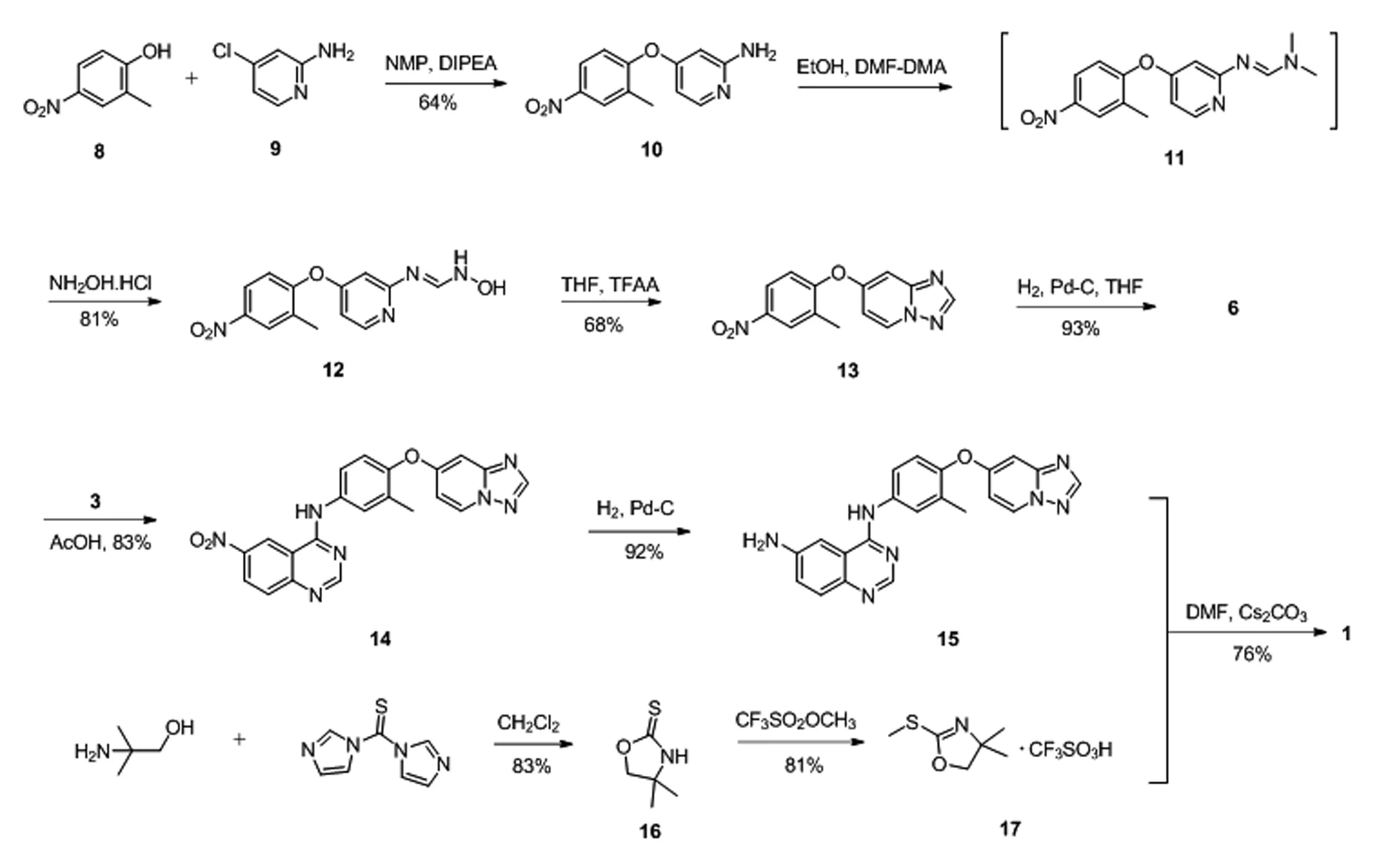
图2 改进的妥卡替尼的合成路线
1 实验部分
1.1 仪器与试剂
实验所用试剂均为市售分析纯,均直接使用,无需进一步纯化;熔点由申光WRS-1B熔点仪测定,温度计未经校正;核磁共振用Bruker UltraShield 400 Plus(TMS为内标)测试;质谱采用Finnigan MAT-95/711测试;HPLC数据采用岛津LC-20AT和Waters 2487 紫外/可见检测器测试。
1.2 试验方法
1.2.1 4-(2-甲基-4-硝基苯氧基)吡啶-2-胺(10)的制备
将N-甲基吡咯烷酮(200 g)、二异丙基乙胺(DIPEA)(67 g,0.52 mol)、2-甲基-4-硝基苯酚(8)(40 g,0.26 mol)和4-氯代-2-氨基吡啶(9)(34 g,0.26 mol)加入2 L三口瓶内,150℃下搅拌48 h,冷却至室温,加水(800 mL)搅拌2 h,抽滤得固体,用水(60 mL×2)洗涤,45℃干燥6 h,得棕色固体(68 g)。将粗产物和活性炭(7 g)放入茄瓶,加THF(300 mL),加热回流2 h,趁热过滤除去固体,将滤液浓缩至约100 g,在室温下搅拌12 h后抽滤收集固体,用THF(20 mL×2)洗涤,在45℃下干燥12 h,得产物10(40.5 g,64%),为灰白色固体。1H NMR (400 MHz, DMSO-d6) δ 8.29 (d, J = 2.6 Hz, 1H), 8.14 (dd, J = 8.9, 2.8 Hz, 1H), 7.88 (d, J = 5.8 Hz, 1H), 7.23 (d, J = 8.9 Hz, 1H), 6.21 (dd, J = 5.8, 2.2 Hz, 1H),6.10 (s, 2H),5.90 (d, J = 2.1 Hz, 1H),2.28 (s, 3H);13C NMR (100 MHz, DMSO-d6): δ 16.1, 95.1, 102.8, 120.9, 123.8, 127.2, 131.8, 144.2, 150.5, 158.5, 162.3, 164.0;MS (ESI):m/z=246.1 [M+H]+。
1.2.2 N-羟基-N'-(4-(2-甲基-4-硝基苯氧基)吡啶-2-基)甲酰胺(12)的制备
将化合物10(39 g,0.159mol)溶于EtOH(200 mL),搅拌,加入DMF-DMA(21 g,0.175 mol),75℃下搅拌3 h;将溶液冷却至~40℃,加入羟胺盐酸盐(13 g,0.19 mol),50℃搅拌3 h,有黄色固体出现,冷却至室温,抽滤后得到黄色固体,用EtOH(20 mL×2)洗涤,45℃下干燥6 h,得到化合物12(37 g,81%),为淡棕褐色固体。1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.37 (d, J = 9.9 Hz, 1H), 8.33 (d, J = 2.3 Hz, 1H), 8.16 (dd, J = 8.9, 2.6 Hz, 1H), 8.10 (d, J = 5.8 Hz, 1H), 7.82 (d, J = 9.9 Hz, 1H), 7.32 (d, J = 10.6 Hz, 1H), 6.61 (d, J = 2.1 Hz, 1H), 6.55 (dd, J = 5.8, 2.2 Hz, 1H), 2.28 (s, 3H);13C NMR (100 MHz, DMSO-d6): δ 16.1, 97.8, 106.5, 121.6, 123.9, 127.3, 132.2, 135.9, 144.7, 150.2, 155.2, 157.9, 164.6;MS (ESI):m/z=289.1 [M+H]+。
1.2.3 7-(2-甲基-4-硝基苯氧基)-[1,2,4]三唑并[1,5-a]吡啶(13)的制备
将化合物12(22 g,77 mmol)溶于THF(200 mL),搅拌,冷却至内温5℃,缓慢滴加TFAA(17 g,80 mmol),控制内温<15℃。滴加完毕后于20~25℃下搅拌反应5 h得澄清溶液,减压除去溶剂,得黄色油状物,加入乙酸乙酯(150 mL),用水(100 mL×2)、饱和NaHCO3(100 mL×1)洗涤有机层,分液,有机相减压除去溶剂,得到淡黄色油状物。加入MeOH(100 mL)、活性炭(2 g),加热搅拌回流1 h,趁热过滤除去固体,滤液在室温下搅拌12 h,抽滤得固体,MeOH(10 mL×2)洗涤,45℃下干燥12 h,得化合物13(14.2 g,68%),为黄色固体。1H NMR (400 MHz, DMSO-d6) δ 9.02 (d, J = 7.4 Hz, 1H), 8.46 (s, 1H), 8.33 (d, J = 2.4 Hz, 1H), 8.13 (dd, J = 8.9, 2.7 Hz, 1H), 7.30 (d, J = 9.0 Hz, 1H), 7.27 (d, J = 2.4 Hz, 1H), 7.10 (dd, J = 7.4, 2.5 Hz, 1H), 2.36 (s, 3H);13C NMR (100 MHz, DMSO-d6): δ 16.1, 101.9, 108.9, 120.2, 123.9, 127.4, 131.4, 131.5, 144.4, 151.2, 154.9, 157.9, 158.6;MS (ESI):m/z= 271.1 [M+H]+。
1.2.4 4-([1,2,4]三唑并[1,5-a]吡啶-7-氧基)-3-甲基苯胺(6)的制备
将化合物13(13.5 g,0.05 mol)和5%湿Pd-C(0.7 g)置于茄瓶,加入THF(120 g),在40℃,用H2气球加氢反应,搅拌15 h,得到澄清棕色溶液,用硅藻土垫过滤,滤饼用THF(10 mL×1)洗涤。合并滤液减压浓缩,得到黄色油状物,加入MeOH(20 mL)在50℃下搅拌,得到澄清溶液,缓慢加入水(20 mL),在室温下搅拌12 h,抽滤得固体,用MeOH(10 mL×2)洗涤,45℃下干燥12 h,得到化合物6(11.2 g,93%),为灰白色固体,mp: 155.5~160.0℃;1H NMR (400 MHz, DMSO-d6) δ 8.87 (d, J = 7.4 Hz, 1H), 8.34 (s, 1H), 6.95 (dd, J = 7.4, 2.1 Hz, 1H), 6.82 (d, J = 8.5 Hz, 1H), 6.63 (d, J = 2.1 Hz, 1H), 6.54 (s, 1H), 6.50 (d, J = 8.4 Hz, 1H), 5.11 (s, 2H), 1.99 (s, 3H);13C NMR (100 MHz, DMSO-d6): δ 16.1, 96.9, 107.6, 113.2, 116.7, 122.3, 130.5, 130.6, 142.1, 147.3, 151.7, 154.9, 161.1;MS (ESI): m/z =241.0 [M+H]+;HPLC归一化法:色谱柱Agilent Eclipse XDB-C18(250 mm×4.6 mm×5 μm);检测波长:254 nm;流速:1.0 mL/min;柱温:35℃;进样量:1 μL;溶剂:MeOH;浓度:0.5 mg/mL;运行时间:20min;流动相:甲醇-水(80: 20);保留时间:4.088 min,纯度:99.13%。
1.2.5 N-(4-([1,2,4]三唑并[1,5-a]吡啶-7-氧基)-3-甲基苯基)-6-硝基喹唑啉-4-胺(14)的制备
根据已报道的方法制备N'-(2-氰基-4-硝基苯基)-N,N-二甲基甲酰亚胺(3),分离提纯后得到88%的淡红色固体[3]。1H NMR (400 MHz, DMSO-d6) δ 8.48 (d, J = 2.4 Hz, 1H), 8.29~8.26 (m, 2H), 7.38 (d, J = 9.2 Hz, 1H), 3.17 (s, 3H), 3.09 (s, 3H);MS (ESI): m/z = 219.1 [M+H]+。
将化合物3(26.2 g,0.12 mol)和化合物6(29.0 g,0.12 mol)置于茄瓶,加入乙酸(250 g),加热至85℃搅拌2 h,得到浅黄色悬浮液,减压蒸馏除去部分溶剂后加水(120 g),在0~5℃下搅拌1 h,抽滤得固体,用水(50 g×2)洗涤,45℃下干燥12 h,得到化合物14(41.2 g,83%),为黄色固体。1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 9.69 (s, 1H), 8.95 (d, J = 7.5 Hz, 1H), 8.75 (s, 1H), 8.57 (dd, J = 9.2, 2.2 Hz, 1H), 8.39 (s, 1H), 7.95 (d, J = 9.2 Hz, 1H), 7.86 (d, J = 11.5 Hz, 2H), 7.25 (d, J = 8.5 Hz, 1H), 7.04 (dd, J = 7.5, 2.5 Hz, 1H), 6.84 (d, J = 2.4 Hz, 1H), 2.22 (s, 3H);13C NMR (100 MHz, DMSO-d6): δ 16.3, 98.3, 107.9, 114.8, 121.2, 121.6, 122.6, 126.2, 127.0, 129.9, 130.4, 130.9, 136.6, 144.9, 148.7, 151.6, 153.5, 155.1, 158.1, 159.1, 159.9;MS (ESI): m/z = 414.1 [M+H]+;HPLC归一化法:色谱柱:Agilent Eclipse XDB-C18(250 mm×4.6 mm×5 μm);检测波长:254 nm;流速:1.0 mL/min;柱温:35℃;进样量:1 μL;溶剂:MeOH;浓度:0.5 mg/mL;运行时间:40 min;流动相:甲醇-水(70∶30);保留时间:23.30 min,纯度:99.47%。
1.2.6N4-(4-([1,2,4]三唑并[1,5-a]吡啶-7-氧基)-3-甲基苯基)喹唑啉-4,6-二胺(15)的制备
在茄瓶里加入化合物14(32.0 g,0.077 mol)、5%湿Pd-C(1.8 g)、THF(240 g),用H2气球加氢,室温搅拌5 h,得到澄清的棕色溶液。用硅藻土垫过滤,滤饼用THF(40 mL×2)洗涤。浓缩合并滤液至约70 g,加水(120 g),在室温下搅拌2 h。抽滤得固体,用水(30 g×2)洗涤,45℃下干燥12 h,得化合物15(27.6 g,92%),为黄色固体。1H NMR (400 MHz, DMSO-d6) δ 9.38 (d, J = 13.8 Hz, 1H), 8.93 (d, J = 7.4 Hz, 1H), 8.38 (s, 1H), 8.36 (s, 1H), 7.90 (d, J = 2.1 Hz, 1H), 7.85 (dd, J = 8.7, 2.3 Hz, 1H), 7.54 (t, J = 8.1 Hz, 1H), 7.38 (d, J = 2.2 Hz, 1H), 7.26 (dd, J = 8.9, 2.2 Hz, 1H), 7.18 (d, J = 8.7 Hz, 1H), 7.03 (dd, J = 7.5, 2.6 Hz, 1H), 6.79 (d, J = 2.4 Hz, 1H), 5.59 (s, 2H), 2.19 (d, J = 5.5 Hz, 3H);13C NMR (100 MHz, DMSO-d6): δ 16.3, 98.3, 107.8, 107.9, 121.3, 121.6, 122.7, 126.3, 127.1, 129.9, 130.5, 130.9, 136.5, 145.0, 148.7, 151.7, 153.5, 155.5, 158.1, 159.2, 159.9;MS (ESI):m/z= 384.1 [M+H]+;HPLC归一化法:色谱柱:Agilent Eclipse XDB-C18(250 mm×4.6 mm×5 μm);检测:254 nm;流速:1.0 mL/min;柱温:35℃;进样量:1 μL;溶剂:MeOH;浓度:0.5 mg/mL;运行时间:40 min;流动相:甲醇-水(70∶30);保留时间:6.475 min,纯度:99.90%。
1.2.7 4,4-二甲基恶唑烷-2-硫酮(16)的制备
将2-氨基-2-甲基-1-丙醇(13.4 g, 0.15 mol)溶于CH2Cl2(200 g)中,缓慢滴加1,1'-(硫代羰基)二咪唑(26.7 g,0.15 mol)溶于CH2Cl2(260 g)的溶液,控温<25℃,滴加完毕后在室温下搅拌17 h,用水(150 g×2)洗涤,减压除去溶剂,得到化合物16(16.3 g,83%),为白色固体[5],直接用于下一步反应。1H NMR (400 MHz, DMSO-d6) δ 10.05 (brs, 1H), 4.27 (s, 2H), 1.26 (s, 6H)。
1.2.8 4,4-二甲基-2-(甲硫基)-4,5-二氢恶唑三氟甲磺酸盐(17)的制备
将化合物16(13.1 g,0.1 mol)溶于CH2Cl2(200 g)中,缓慢滴加三氟甲磺酸甲酯(18.0 g,0.11 mol),控温<25℃。滴加完毕后在室温下搅拌15 h。用甲基叔丁基醚(500mL)稀释反应液,在0~5℃下搅拌2 h。抽滤得固体,用甲基叔丁基醚(30 mL×2)洗涤,室温下真空干燥6 h,得化合物17(23.9 g,81%),为白色固体,直接用于下一步反应。1H NMR (400 MHz, DMSO-d6) δ 4.56 (s, 2H), 2.61 (s, 3H), 1.39 (s, 6H)。
1.2.9 妥卡替尼(1)的制备
将化合物17(16.2 g,55 mmol)溶于DMF(80 mL)中,室温下将Cs2CO3(26.5 g,82 mmol)分批加入其中,搅拌反应1 h后加入化合物15(16.9 g,44 mmol),125℃下搅拌20 h。冷却至室温加入冰水(300 g),室温搅拌1 h。抽滤得固体,用水(20 mL×2)洗涤,在45℃下干燥6 h,得到浅黄色固体21 g。将粗产物和活性炭(4 g)置于EtOAc(100 mL)中加热回流1 h。趁热过滤除去固体,将滤液浓缩至约50 g,将正庚烷(50 mL)缓慢加至温热的溶液中,在室温下搅拌12 h。抽滤得固体,用正庚烷(15 mL×2)洗涤,45℃下干燥12 h,得到化合物1(16.1 g,76%),为白色固体,mp为251.2~254.7℃。1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 1H), 8.94 (d, J = 7.5 Hz, 1H), 8.50 (s, 1H), 8.38 (s, 1H), 8.03 (brs, 1H), 7.92 (s, 1H), 7.87 (d, J = 8.5 Hz, 1H), 7.67 (d, J = 8.5 Hz, 1H), 7.59~7.41 (m, 1H), 7.20 (d, J = 8.7 Hz, 1H), 7.03 (dd, J = 7.5, 2.6 Hz, 1H), 6.80 (d, J = 2.3 Hz, 1H), 4.08 (s, 2H), 2.19 (s, 3H), 1.29 (s, 6H);13C NMR (100 MHz, DMSO-d6): δ 16.3, 27.5, 78.1, 97.9, 107.8, 116.2, 121.6, 121.9, 125.4, 128.5, 130.2, 130.8, 137.9, 145.9, 147.7, 151.7, 152.6, 155.1, 157.4, 160.2;MS (ESI): m/z =481.2 [M+H]+;HPLC归一化法:色谱柱:Agilent Eclipse XDB-C18(250 mm×4.6 mm×5 μm);检测波长:254 nm;流速:1.0 mL/min;柱温:35℃;进样量:1 μL;溶剂:MeOH;浓度:0.5 mg/mL;运行时间:40 min;流动相:甲醇-水(70∶30);保留时间:7.563 min,纯度:99.9%。
2 总结与讨论
本文主要开发了妥卡替尼(1)的新工艺路线并对其进行改进,重点合成其三个关建中间体,其中:4-([1,2,4]三唑并[1,5-a]吡啶-7-氧基)-3-甲基苯胺(6)经5步反应制得,产率32.8%,纯度99.1%;N4-(4-([1,2,4]三唑并[1,5-a]吡啶-7-氧基)-3-甲基苯基)喹唑啉-4,6-二胺(15)经3步反应制得,产率67.0%,纯度99.5%;4,4-二甲基-2-(甲硫基)-4,5-二氢恶唑三氟甲磺酸盐(17)经2步反应制得,产率为67.2%;最后化合物15和17反应制得化合物1,产率76%,纯度>99.5%。
在由2-甲基-4-硝基苯酚(8)和4-氯吡啶-2-胺(9)制备4-(2-甲基-4-硝基苯氧基)吡啶-2-胺(10)的步骤中,分离纯化后的收率~64%,造成收率不高的原因目前认为是在后处理过程中损耗较大,使用活性炭热滤、重结晶操作影响了收率,但通过TLC和HPLC的监测未发现有明显杂质,由于化合物10的合成不是本工艺路线的关建步骤,因此目前还未对该步反应优化。
本文中采用了H2/Pd-C的催化氢化的反应条件将苯环上硝基还原成氨基,分别由化合物13制备6、化合物14制备15,因为这几个化合物中均含有三氮唑结构,为了避免碳氮双键被还原,我们均采用氢气球加氢、室温~40 ℃相对温和的反应条件,实验分析结果表明该催化氢化反应收率较高,未发现有副产物生成。
恶唑烷-2-硫酮化合物16和甲硫基二氢恶唑化合物17是基于文献报道的方法经优化后制备[5-6],这两个化合物均较不稳定,需要现用现制,室温存放溶解分解变质。其中化合物16相对于17要稳定一些,在4 ℃下可以储存2~3周不变质,而化合物17则需在-18℃,Ar2的保护下储存才能保证不变质。
综上所述,本论文成功制备了抗肿瘤药物妥卡替尼,并对该新工艺路线进行了初步优化,经9步反应制得妥卡替尼,总收率16.7%,纯度>99.5% (HPLC),原料试剂易得、反应条件温和、操作安全方便,目标产物及中间体结构经1H-NMR、13C-NMR和LC-MS确证。
猜你喜欢
云南化工(2022年9期)2022-10-12
安徽化工(2022年1期)2022-02-15
能源化工(2021年6期)2021-02-26
中华养生保健(2020年3期)2020-11-16
化工学报(2020年4期)2020-05-28
今日农业(2019年11期)2019-08-13
铜仁学院学报(2018年6期)2018-07-05
中国洗涤用品工业(2016年2期)2016-02-28
中国塑料(2015年2期)2015-10-14
云南中医学院学报(2015年2期)2015-07-31
