
基于pH响应两亲性卟啉嵌段共聚物的光动力与化学联合治疗
2020-05-07 12:04张显杨王廷虎许立人顾邯沙张伟安
华东理工大学学报(自然科学版) 2020年2期
肖 潮, 田 佳, 刘 峰, 张显杨, 王廷虎, 许立人, 顾邯沙, 张伟安
(华东理工大学材料科学与工程学院,上海 200237)
近年来癌症的发病率不断上升,由于没有特别有效的治愈方法,死亡率一直居高不下[1]。光动力疗法(PDT)被希冀于成为一种有效的癌症治疗手段,该疗法是通过先将光敏剂(PS)聚集在肿瘤部位,然后PS在相应的激光照射下,产生高能量的活性氧(ROS),从而杀死癌细胞[2-4]。相比化疗、激光治疗和手术切除等目前常用的治疗方法,PDT具有可控、局部定位、副作用小、治疗彻底等优势[5-7]。一些光敏剂目前已经得到美国食品药品局(FDA)的认可,其中基于维替泊芬(Verteporfin)的药剂维速达尔已经大量用于治疗黄褐斑与病理性近视[8]。四苯基卟啉(TPP)作为第一代光敏剂,近年来一直是PDT的热点研究对象,但其单线态氧产率不高、靶向性差、小分子水溶性差、化学修饰困难、生物相容性差和易聚集引起猝灭等缺点一直是阻碍其发展和临床应用的难点[9-15]。现阶段最常用的方式是直接将卟啉物理包封或包覆在一个纳米药物运送系统中,然后借助于大分子纳米颗粒的生物相容性、实体瘤的高通透性和滞留(EPR)效应等优点,将卟啉运送到肿瘤部位,再在肿瘤部位进行光照,从而实现肿瘤部位的局部治疗[16-23]。Li等[24]将四苯基卟啉掺杂在以聚(苯乙烯-马来酸酐)和聚[(9,9-二辛基芴-2,7-二基)-交替-(2,2'-联二噻吩-5,5'-二基)]制成的纳米量子点中,然后以聚合物的纳米量子点为载体,以解决其生物相容性差、化学修饰困难等缺点,实现了较好的肿瘤治疗效果。Roy等[25]将三乙氧基乙烯基硅烷水解制成尺寸为30 nm左右的二氧化硅纳米颗粒,并将卟啉直接物理包封其中,通过EPR效应,二氧化硅纳米颗粒可以将卟啉运载到肿瘤部位,以解决其在水溶液中易聚集、生物相容性差、化学修饰困难等缺点。Ding等[26]则合成了两种特殊的卟啉分子,将其直接制成尺寸合适的纳米颗粒,以解决卟啉水溶性差、易聚集的缺点;并利用EPR效应,使纳米颗粒在肿瘤处聚集,从而实现了较好的肿瘤治疗效果[26]。以上通过物理包载的方式将TPP运送到肿瘤部位的方法,虽然解决了卟啉的一些固有缺点,但依旧存在一些不足之处:(1)小分子卟啉在肿瘤细胞处释放出来后,依旧十分容易聚集猝灭,导致单线态氧产率的降低;(2)卟啉只是简单地物理包载在纳米颗粒内部,在生物体内长循环时容易泄露,从而使得肿瘤部位的卟啉量降低[27]。因此,构建具有肿瘤内源性响应的双亲性的卟啉聚合物不仅能够解决药物提前泄露问题,还能减少卟啉堆积引起的自淬灭现象。
本文结合可逆加成-断裂链转移(RAFT)聚合与点击化学偶联反应构建了一种两亲性嵌段共聚物聚乙二醇-聚甲基丙烯酸二异丙基氨基乙酯-四苯基卟啉(PEG-PDPA-TPP),该共聚物的中间连接点上有一个可用作光动力疗法中光敏剂的四苯基卟啉(TPP)基元。首先通过RAFT聚合制备了中间节点为叠氮的双亲性聚合物PEG-PDPA-N3和带有炔基官能团的TPP,然后利用点击化学将TPP偶联到亲水段与疏水段的连接点上。这种两亲性的嵌段共聚物由亲水性的聚乙二醇(PEG)嵌段、疏水性的聚甲基丙烯酸二异丙基氨基乙酯(PDPA)嵌段和连接点上的TPP组成。卟啉通过共价化学键相连在大分子PEGPDPA上,使得TPP能够稳定地固定在纳米颗粒内,从而解决其在生物体内长循环时易泄露的问题。并且PEG-PDPA-TPP在水溶液中组装形成包载盐酸阿霉素(DOX)的纳米胶束,可进一步与化疗联合治疗癌症。因此PEG-PDPA-TPP不仅是修饰了TPP的大分子光敏剂,同时也是化疗药物DOX极佳的运载和释放载体,能够在癌细胞内的酸性条件下释放出DOX。最重要的是,通过分子设计将TPP单元偶联在亲水段和疏水段中间点使得TPP在疏水和亲水组分之间规则排列而不是聚集在一起,从而解决了卟啉聚集引起的单线态氧产率下降的问题,提高了大分子光敏剂PEG-PDPA-TPP的光动力效果。因此,这种装载DOX的基于pH响应的含TPP嵌段共聚物的药物释放平台能够解决卟啉在生物癌症治疗上的一些问题,为癌症治疗提供一种新策略。
1 实验部分
1.1 原料和试剂
聚乙二醇单甲醚(PEG,Mn= 5 000):98%(纯度,下同),Sigma-Aldrich有限公司;叠氮化钠(NaN3):97%,阿拉丁试剂有限公司;氢化钠(NaH):99%,阿拉丁试剂有限公司;表氯醇:98%,阿拉丁试剂有限公司;二环己基碳二亚胺(DCC):97%,阿拉丁试剂有限公司;4-二甲基-氨基吡啶(DMAP):97%,阿拉丁试剂有限公司;五甲基二亚乙基三胺(PMDETA):98%,泰坦科技有限公司;溴化亚铜(CuBr):98%,泰坦科技有限公司;甲基丙烯酸二异丙基氨基乙酯(DPA):98%,泰坦科技有限公司;溴丙炔:98%,泰坦科技有限公司;1,3-二苯基异苯并呋喃(DPBF):98%,泰坦科技有限公司;偶氮二异丁腈(AIBN):98%,泰坦科技有限公司;盐酸阿霉素(DOX·HCl):97%,北京华丰联合技术公司;Hochest33342(核酸染料):98%,碧云天生物技术有限公司;3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐(MTT):98%,碧云天生物技术有限公司;甲苯,1,4-二氧六环,浓盐酸(HCl),甲醇(MeOH),二氯甲烷(DCM),四氢呋喃(THF),N,N-二甲基甲酰胺(DMF)和三乙胺(TEA)等溶剂购自泰坦科技有限公司(分析纯)并且在使用前加入氢化钙干燥蒸馏收集。参照已发表文献的方法合成了链转移剂(CTA)4-氰基-4-(十二烷基硫基硫代羰基)硫烷基戊酸(CDSPA)[28]。羟基化四苯基卟啉5-(4-Hydroxylphenyl)-10,15,20-triphenyl-porphyrin (TPP-OH)参照文献[4]合成。
1.2 测试与表征
氢核磁共振仪(400 MHz,德国Brucker公司,AVANCE Ш 500型):在室温下测量,以四甲基硅烷(TMS)为内标,氘代氯仿为溶剂。
紫外-可见光光度计(日本SHIMADZU公司,UV-2550型):室温下,样品溶液在石英比色皿中检测,扫描范围300~900 nm。
纳米粒度仪(美国Beckman Coulter Delsa Nano C型):测试标样为水相,测试范围0.6~7 000.0 nm。
共聚焦仪(日本尼康公司,AIR型):水相60倍放大,激发光波长420 nm,接收范围630~675 nm。
1.3 实验过程
1.3.1 环氧化物封端的PEG(PEG-epoxy)的合成 将PEG(10.0 g)溶解在80 mL无水甲苯中。共沸蒸馏掉20 mL甲苯后,在氮气环境保护下将NaH(1.0 g,25 mmol)加入到溶液中。在35 ℃油浴锅中搅拌24 h后,将环氧氯丙烷(4.0 mL,50 mmol)加入到溶液中,40 ℃下搅拌24 h。之后通过离心除去反应中所产生的无机盐,并将上清液在旋转蒸发器上浓缩。将聚合物溶解在少量DCM中并在冰乙醚中沉淀2次,过滤并真空干燥后,收集8.6 g PEG-epoxy。1H-NMR(400 MHz,Chloroform-d,δ) 3.48~3.82 (b,456H),3.31(s,3H),3.10 (d,1H), 2.73 (d,1H),2.54 (d,1H)。
1.3.2 α,α'-叠氮-羟基-PEG(PEG-OH-N3)的合成 PEG-OH-N3通过环氧化物封端的PEG与NaN3的反应合成。将PEG-epoxy(2.0 g,0.38 mmol)溶解在25 mL DMF中,并 将NaN3(0.13 g,2.0 mmol)和NH4Cl(0.11 g,2.0 mmol)混合加入到溶液中。60 ℃下搅拌60 h后,在旋转蒸发器上除去溶剂,然后用DCM稀释并用饱和食盐水洗涤3次。将收集到的有机相用无水硫酸镁干燥,过滤并在旋转蒸发器上浓缩。将残余物在冰乙醚中沉淀3次,过滤并真空干燥后,得 到1.85 g PEG-OH-N3。1H-NMR (400 MHz,Chloroform-d,δ) 3.95 (s,1H),3.61~3.73 (s,458H),3.38(s,3H),3.34 (d,2H)。
1.3.3 酯化反应合成PEG-CDSPA-N3将PEG-OHN3(1.0 g,0.2 mmol)溶解在25 mL无水DCM中,将CDSPA(0.22 g,0.6 mmol)和DMAP(24.4 mg,0.2 mol)加入该溶液中,然后,在氮气环境下冰浴滴加DCC(51.5 mg,0.25 mmol)的DCM溶液。在25 ℃下搅拌24 h后,用旋转蒸发器除去溶剂,然后用DCM稀释并用饱和食盐水洗涤3次。将收集到的有机相用无水硫酸镁干燥,过滤并在旋转蒸发器上浓缩。将残余物在冰乙醚中沉淀3次,过滤并干燥后,得到0.86 g PEG-CDSPA-N3。1H-NMR (400 MHz,Chloroform-d,δ) 3.96 (s,3H),3.59~3.71 (s,458H),3.38 (s,3H),3.34(d,4H),2.26 (t,2H),1.26 (s,20H),0.88 (t,3H)。
1.3.4 RAFT聚合制备PEG-PDPA-N3将PEG-CDSPA-N3(0.1 g,0.018 mmol)、DPA(0.45 g,2.3 mmol)、AIBN(1.0 mg,0.006 mmol)和1.5 mL 1, 4-二氧六环加入Schlenk瓶中。经过3次冻融循环除氧后在70 ℃下搅拌18 h,之后将粗产物溶解在甲醇中并用甲醇和水(体积比3∶1)的混合溶液透析以除去未反应的单体和副产物,然后用去离子水进一步透析以除去甲醇(透析膜截留分子量(MWCO)为8 000)。最后经过2 d的冻干后得到0.39 g 聚合度为46的产物PEGPDPA-N3。1H-NMR (400 MHz,Chloroform-d, δ) 3.99(d, 92H),3.65 (s,458H),3.38 (s, 3H),2.95 (d,92H),2.56~2.76 (m,92H), 1.67~1.84 (m,96H),0.81~1.21 (m,646H)。
1.3.5 以炔基为单官能团的卟啉(TPP(Zn2+)-yne)的合成 将单羟基四苯基卟啉锌配合物(1.0 g,1.63 mmol)溶解于30 mL的无水DCM中,然后加入溴丙炔(387.9 mg,3.31 mmol)和K2CO3(164.6 mg,1.63 mol),之后在氮气保护下,40 ℃油浴锅中反应24 h。反应结束后,用饱和食盐水洗涤3次,旋干溶剂。留下的固体过硅胶柱分离,洗脱剂为乙酸乙酯和石油醚(体积比1∶3)的混合溶液,最后将收集到的目标组分旋干并真空干燥,得到0.76 g 的炔基卟啉(TPP(Zn2+)-yne)。1H-NMR (400 MHz, Chloroform-d, δ) 8.87 (d,8H),8.17 (m,6H),8.09 (d,2H), 7.64 (m,9H),7.32 (d,2H),4.91 (s,2H),2.62 (s,1H)。
1.3.6 通过点击化学反应合成PEG-PDPA-TPP 将炔基官能化的四苯基卟啉(10.4 mg,0.016 mmol)和PEG-PDPA-N3(201.2 mg,0.013 mmol)溶于30 mL无水THF中,通氮气除氧40 min后加入CuBr(3 mg,0.018 mmol)和PMDETA(3.5 mg,0.02 mmol),然后再次通氮气20 min。在50 ℃油浴锅中搅拌48 h,结束后旋干溶剂并溶于MeOH与DCM(体积比1∶3)的混合溶剂中,再加入0.1 mL浓盐酸,室温搅拌6 h后旋干溶剂,用饱和食盐水洗3次后旋干,然后将粗产物溶解在甲醇中并用甲醇和水(体积比3∶1)的混合液透析以除去Cu2+和过量的卟啉,然后用去离子水透析以除去甲醇(MWCO为8 000)。冷冻干燥后,收集到0.16 g 聚合度为46的PEG-PDPA-TPP。1HNMR (400 MHz,Chloroform-d, δ) 8.89 (d,8H),8.21(m,8H),7.76 (m,10H),3.95 (d,93H),3.65 (s,458H),3.38 (s,3H),2.86 (m,92H),2.23 (t,92H),1.67 (m,92H),1.41~1.54 (t,654H)。
TPP修饰的两亲性嵌段共聚物总的合成过程如图1所示。
1.3.7 制备包载DOX和不含DOX的胶束 通过膜透析方法制备单独胶束和包载DOX的胶束。对于单独胶束,将10 mg共聚物溶解在2 mLTHF与DMF(体积比1∶4)的混合溶剂中。磁力搅拌下在30 min内将混合物滴加到3 mL去离子水中。然后用去离子水透析24 h以除去有机溶剂。装载DOX的胶束的制备方法与单独胶束的制备方法相似,如图2所示。将DOX·HCl(1.33 mg,用3倍的TEA处理)和共聚物(10 mg)溶解在2 mL THF与DMF(体积比2∶3)混合溶剂中。在磁力搅拌下将混合物滴加到3 mL去离子水中30 min,用去离子水透析24 h以除去溶剂和游离DOX。冷冻干燥后,将装载DOX的胶束溶解在DMF中,并根据校准曲线通过紫外-可见光光度计在500 nm处测定DOX的量。药物负载量(DLC)定义为包载在胶束中的药物质量与包载药物后胶束的总质量之比,药物包载率(EE)定义为包载在胶束中的药物质量与包载药物质量之比。
1.3.8 临界胶束浓度(CMC)的测定 使用芘作为荧光探针来测定CMC。将芘溶解在丙酮中制备一定浓度的溶液,然后向每个容量瓶中加入一定量的芘溶液和不同浓度的聚合物样品溶液,此时各容量瓶中配制的芘的浓度均为6×10-7mol/L。以335 nm的光激发,接收的荧光波长范围调整为335~450 nm。激发发射狭缝设定为20 nm和10 nm。以383 nm处的荧光吸收强度(I383)和372 nm处的荧光吸收强度(I372)的比值计算胶束的CMC。
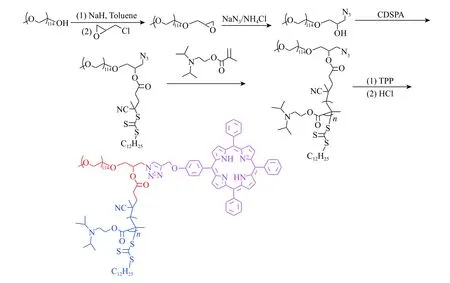
图 1 TPP修饰的两亲性嵌段共聚物的合成步骤Fig. 1 Synthesis procedure of TPP modified amphiphilic block copolymer

图 2 包载DOX的PEG-PDPA-TPP胶束的制备及胶束包吞入细胞内的过程示意图Fig. 2 Schematic diagram showing the preparation of DOXloaded PEG-PDPA-TPP micelles and their encapsulation by cells
1.3.9 体外DOX的释放实验 使用透析膜(截留分子量3 500)研究DOX的释放行为。将DOX装入到透析膜中并浸入到30 mL PBS溶液中,温度保持在37 ℃,转速为100 r/min。每隔一段时间取出1.5 mL透析溶液并加入到等体积的新鲜PBS溶液中。测试500 nm处的紫外吸收强度,再与DOX的标准曲线对比计算DOX的释放速率。
1.3.10 细胞培养实验 将Hela细胞(Henrietta Lacks细胞)在含有w=10%胎牛血清(FBS)和w=1%抗生素(50 U/ mL青霉素和50 U/ mL链霉素)的DMEM培养基中培养。细胞皿放在专门的恒温箱中培养(37 ℃,φ=5% CO2)。
1.3.11 细胞毒性实验 将含有Hela细胞的悬浮液加入到96孔板中孵育,密度为每孔5×103个细胞。对于细胞暗毒实验,在孔板中加入各种浓度的自由DOX、自由TPP、PEG-PDPA-TPP胶束和装载DOX的PEG-PDPA-TPP胶束处理细胞。将细胞放在黑暗中培养24 h,采用标准MTT(5 mg/mL)测定法进行死活测试,使用SpectraMax光谱仪测定560 nm处的吸光度。细胞存活率计算如下:

其中:ODtest是加入药品后的吸光度;ODcontrol是空白对照样的吸光度;ODblank是空板的吸光度。
用不同浓度的PEG-PDPA-TPP、包载DOX的PEG-PDPA-TPP、包载TPP的PEG-PDPA胶束溶液处理Hela细胞并孵育24 h,然后用655 nm(0.3 W/cm2)的激光照射孔板10 min。而后再培养24 h,采用标准MTT(5 mg/mL)测定法评估细胞死活率。
1.3.12 激光共聚焦扫描显微镜(CLSM)成像实验
将约5×103个Hela细胞接种到每个玻璃底的器皿中,并加入含有w=10%FBS的DMEM中培育24 h。然后,用TPP质量浓度为1.0 μg/mL的游离TPP和包载DOX的PEG-PDPA-TPP胶束分别处理细胞,分别培育4 h和24 h后,除去培养基并用PBS溶液洗涤3次,之后将细胞用多聚甲醛(w=4%)的PBS溶液固定25 min,然后用Hoechst 33342染色以标记细胞核。最后,使用荧光显微镜(Nikon AIR)进行细胞的CLSM成像拍摄。
2 结果与讨论
2.1 PEG-PDPA-TPP嵌段共聚物的合成及其表征
通过RAFT聚合和点击化学反应的结合,合成了含TPP的嵌段共聚物。TPP位于亲水性的PEG嵌段和疏水性的PDPA嵌段之间的连接点处(图1)。而后通过自组装形成包载化疗药物DOX的纳米胶束,细胞实验验证其治疗效果。
PEG-PDPA-TPP嵌段共聚物的合成包括以下步骤:
(1)通过将表氯醇接枝到具有单羟基端基的PEG上来合成PEG-环氧树脂。
(2)环氧化物官能化的PRG进一步与叠氮化钠反应,打开环氧环,得到叠氮化物官能化的PEG(PEGOH-N3)。值得注意的是,开环反应同时会产生一个末端羟基。
(3)在室温下通过DCC/DMAP的酯化体系将用于RAFT聚合的链转移剂CDSPA成功引入到PEG链上。将连接了CDSPA的叠氮化物官能化的PEG命名为PEG-CDSPA-N3。
(4)用PEG-CDSPA-N3作为大分子的RAFT剂,通过DPA的RAFT聚合合成嵌段共聚物PEGPDPA-N3。图3(a)示出了PEG-PDPA-N3的1H-NMR谱和各个峰的分配。基于1H-NMR的数据可以计算PDPA的聚合度(DP)为46,同时在其13C-NMR谱(图3(d))中,可以清楚地看到PEG与PDPA链段上的各个碳的归属情况,亦证明了PEG-PDPA-N3被成功合成。
(5)将参照之前文献合成的羟基卟啉与丙炔酸进行酯化反应,合成端基为炔基的卟啉。其氢核磁谱图如图3(b)所示,可以证明其被成功合成。
(6)通过Cu(I)催化叠氮化物与炔烃环的加成反应(点击化学反应)将TPP接到连接点上。图3(c)示出了连接了TPP的嵌段共聚物的1H-NMR谱,其被命名为PEG-PDPA-TPP。同时,将PEG-PDPA-TPP的13C-NMR谱图与PEG-PDPA-N3进行对比,可以清楚地看到TPP苯环上的C特征峰出现在了谱图上(图3(e)中的j和k),亦证明了TPP成功地接在了聚合物上。
体积排除色谱(SEC)用于表征合成聚合物的分子量和多分散性指数(PDI)的变化。官能化的PEG链和嵌段共聚物的SEC曲线如图4所示。PEGCDSPA-N3相对于前一步化合物仅导致SEC曲线产生了轻微偏移。而DPA的RAFT聚合导致SEC线中的洗脱时间的明显减少。连接TPP后,聚合物分子量增大,SEC出峰向左产生了轻微偏移。同时,所有含TPP的嵌段共聚物的分子量分布非常窄。关于聚合物的分子量和分子量分布的详细信息如表1所示。
2.2 PEG-PDPA-TPP等样品在水溶液中的自组装
PEG-PDPA-TPP嵌段共聚物组装后在去离子水中透析后可形成胶束。采用芘用作荧光探针,基于在383 nm和372 nm处的芘单体发射的电子振动带的强度比来测量PEG-PDPA-TPP嵌段共聚物的CMC。随着样品质量浓度的增加,I383/I372的值在低质量浓度时仅略有增加,但在一定质量浓度后呈现显著增加,表明芘分子从水溶液转移到形成的胶束的疏水核心区域。CMC被确定为低质量浓度区域和高质量浓度区域的I383/I372的比率外推的交叉点(图5(a))。同时基于自组装和pH响应行为,PEGPDPA-TPP被用作药物释放的独特载体。选择DOX作为模型化疗药物,并通过纳米沉淀法制备负载DOX的聚合物胶束。将包载DOX的聚合物胶束在去离子水中透析24 h。冻干后溶解于DMF中,根据500 nm处的吸光度计算DOX含量。当DOX与共聚物的质量比为1∶5时,包载有DOX的PEGPDPA-TPP胶束的载药量和包封率分别为3.1%±0.5%和18.6%±3.3%。动态光散射仪(DLS)用于分析PEG-PDPA-TPP、包载DOX的PEG-PDPA-TPP和包载TPP的PEG-PDPA的自组装的尺寸(图5(b))。值得注意的是,所有胶束都具有较窄的尺寸分布。
2.3 包载DOX的PEG-PDPA-TPP胶束的pH响应实验
据报道肿瘤细胞溶酶体中的pH为5.0~5.5。pH响应性的载体需要高度满足在弱酸性的肿瘤细胞微环境下药物释放的要求。PDPA在重复单元中具有叔胺基团,这使其成为pH响应聚合物的理想候选材料。PDPA链在中性或碱性环境下疏水段塌陷,而由于叔胺基团在酸性条件下质子化转变为带电物质,从而会在酸性环境下伸展。采用DLS技术研究包载DOX的PEG-PDPA-TPP胶束在不同pH缓冲溶液中自组装的流体动力学尺寸变化。如图5(b)所示,当pH从7.4降至6.5时,胶束的流体动力学直径从约98 nm增长至378 nm。当缓冲溶液的pH进一步降低至5.5时,组装体的流体动力学尺寸上升至1 081 nm,与此同时PDI呈现上涨,胶束粒径表现出强烈的不稳定性,说明胶束解离并且包载DOX释放值之后聚集。这些结果说明了PEG-PDPA-TPP胶束在pH梯度下的pH响应行为。

图 3 (a)PEG-PDPA-N3,(b)TPP(Zn2+)-yne,(c)PEG-PDPA-TPP的1H-NMR谱图;(d)PEG-PDPA-N3,(e)PEG-PDPA-TPP的13CNMR 谱图Fig. 3 1H-NMR spectra of (a) PEG-PDPA-N3, (b) TPP(Zn2+)-yne, (c)PEG-PDPA-TPP, 13C-NMR spectra of (d) PEG-PDPA-N3 and (e) PEGPDPA-TPP
与此同时,用酸性缓冲溶液模拟细胞内的酸环境,测试包载DOX的PEG-PDPA-TPP胶束在不同pH时的释放行为( 图6)。在pH=7.4的缓冲溶液中,在50 h内从胶束中发现DOX的低释放(21%),这表明胶束在血液循环期间相对稳定。然而,当缓冲溶液的pH降至5.5时,由于胶束的解离,有72%的DOX释放。这表明聚合物胶束是DOX释放的良好载体,而它在血液循环期间是稳定的,但在细胞摄取后能够有效释放DOX。
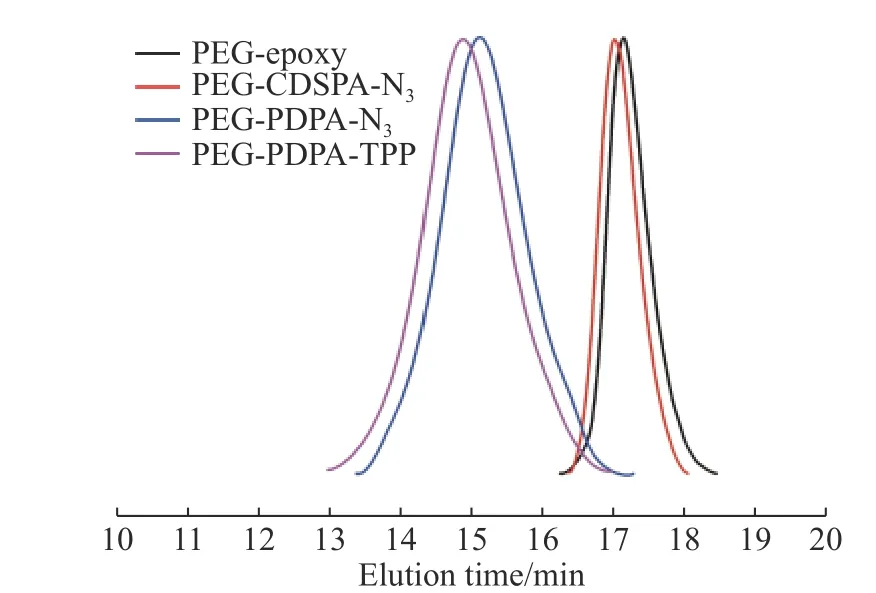
图 4 PEG-epoxy、 PEG-CDSPA-N3、 PEG-PDPA-N3,和PEG-PDPA-TPP的体积排除色谱曲线Fig. 4 SEC traces of PEG-epoxy, PEG-CDSPA-N3, PEG-PDPAN3 and PEG-PDPA-TPP

表 1 聚合物的分子量和多分散性Table 1 Molecular weight and polydispersity of the polymers
2.4 聚合物单线态氧产生实验
卟啉作为一种典型的光敏剂,在癌症治疗方面有很好的研究,然而由于卟啉的官能化困难,难以连接到聚合物上,因此通常只是包裹在纳米载体中,而TPP的聚集则会引起其单线态氧产率的降低。将DBPF用作猝灭剂以评估PEG-PDPA-TPP胶束的单线态氧量子产率。通过UV-vis吸收监测单线态氧猝灭的DPBF的量。测定了TPP(含w=0.1%DMSO的水溶液)和PEG-PDPA-TPP胶束在不同655 nm激光照射时间下的UV-vis光谱变化。为了获得更直观的结果,收集样品在410 nm处的吸收强度值(I410)并绘制在图7中。TPP样品显示出较低的单线态氧量子产率的原因主要是因为TPP的聚集。相反,在PEGPDPA-TPP胶束中,由于TPP组装在球形胶束的表面,减少了TPP的聚集并且拥有确定的形态,从而具有更高的单线态氧量子产率。因此,在同等TPP浓度下,PEG-PDPA-TPP胶束使得TPP的聚集猝灭效应减少,其单线态氧产率会明显高于自由的TPP,表现在测试结果中则是PEG-PDPA-TPP 胶束中的DPBF的吸收峰的衰减速度会明显快于TPP,这与实验结果相符合。

图 5 (a)PEG-PDPA-TPP和PEG-PDPA的临界胶束浓度;(b)动态光散射仪测试的PEG-PDPA + TPP,PEG-PDPA-TPP,PEGPDPA-TPP + DOX(PBS,pH=7.4),PEG-PDPA-TPP + DOX(pH=6.5)和PEG-PDPA-TPP + DOX(pH=5.5)粒径分布图Fig. 5 (a) CMC of PEG-PDPA-TPP and PEG-PDPA (b) Hydrodynamic diameters of PEG-PDPA + TPP, PEG-PDPA-TPP, PEG-PDPA-TPP +DOX (PBS, pH=7.4), PEG-PDPA-TPP + DOX (pH=6.5); and PEG-PDPA-TPP + DOX (pH=5.5) micelles in aqueous solution determined by DLS
2.5 细胞毒性实验
通过MTT测定法研究了PEG-PDPA-TPP胶束、包载DOX的PEG-PDPA-TPP胶束、游离DOX和游离TPP细胞的暗毒,结果如图8(a)所示。由于PEG链段和TPP具有良好的生物相容性,因此嵌段共聚物PEG-PDPA-TPP和游离TPP几乎不显示细胞暗毒。另一方面,通过增加样品浓度,游离DOX的细胞增殖抑制显著增加。值得注意的是,包载DOX的PEG-PDPA-TPP胶束显示出比PEG-PDPA-TPP更高的细胞暗毒,但是比游离DOX的细胞暗毒低得多。这主要是由于pH响应性PDPA链在细胞摄取后在酸性微环境中会有DOX的缓释作用,相比于游离DOX要温和许多。表明包载DOX的PEG-PDPATPP胶束是良好的药物释放平台。
为了分析PEG-PDPA-TPP胶束和包载TPP的PEG-PDPA胶束之间的差异,进一步研究了具有相同TPP含量的两种样品的光毒性(图8(b))。由暗毒实验可知两种样品几乎都没有暗毒性。使用激光束(655 nm,300 mW/cm2)照射孔板10 min从而进一步研究样品的光毒性。PEG-PDPA-TPP胶束在光毒性方面显示出比包载TPP的PEG-PDPA胶束更高的细胞增殖抑制作用,即更大的细胞毒性。这表明在中间节点处连接有TPP的嵌段共聚物表现出比物理包封更显著的癌症治疗功效,这与单线态氧量子产率的结果高度一致。同时包载DOX的PEG-PDPATPP胶束在655 nm激光照射(10 min)后相比于PEGPDPA-TPP(光毒)和PEG-PDPA-TPP+DOX胶束(暗毒)展现出更好的细胞抑制作用,表明化疗与光动力治疗相结合之后,包载DOX的PEG-PDPA-TPP胶束具有更佳的癌细胞抑制效果。
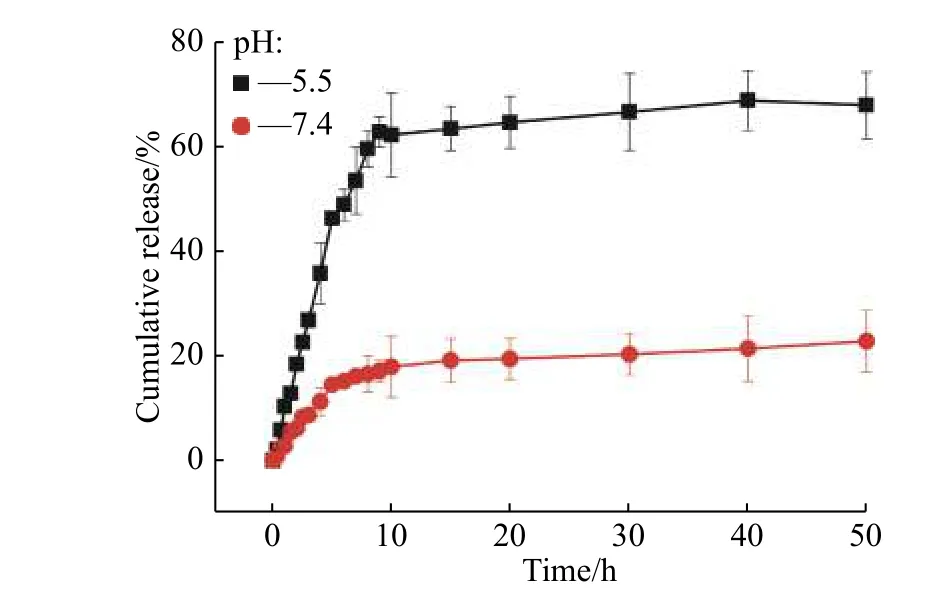
图 6 包载有DOX的PEG-PDPA-TPP胶束在不同pH的磷酸盐缓冲液中的体外释放行为(37 °C)Fig. 6 In vitro release behavior of DOX-loaded PEG-PDPA-TPP micelles in PBS buffer solutions with different pH ( 37°C)

图 7 PEG-PDPA-TPP胶束和纯TPP(w=0.1%的DMSO)加入DPBF后,随着激光照射(655 nm,0.3 W/cm2,ρTPP=3.0 μg/mL)不同时间后,在410 nm处的吸光强度相对衰减图Fig. 7 Plots of I420 against irradiation time, PEG-PDPA-TPP micelles with DPBF probe and free TPP (w =0.1% DMSO)micelles with DPBF probe (655 nm, 0.3 W/cm2, ρTPP=3.0 μg/mL)

图 8 用MTT法测试的细胞存活率:(a)PEG-PDPA-TPP、包载的 PEG-PDPA-TPP+DOX胶束、游离DOX和游离TPP的暗毒;(b)PEG-PDPA-TPP 胶束、 PEG-PDPA+TPP胶束的光毒和暗毒、PEG-PDPA-TPP + DOX胶束的暗毒和光毒(655 nm,300 mW/cm2,10 min)Fig. 8 Cell viability determined by MTT assay: (a) Dark toxicity of PEG-PDPA-TPP micelles, PEG-PDPA-TPP+DOX micelles, free DOX and free TPP; (b) Photo toxicity and dark toxicity of PEG-PDPA-TPP micelles, PEG-PDPA+TPP micelles,dark and photo toxicity of PEGPDPA-TPP + DOX micelles (655 nm, 300 mW/cm2, 10 min)
2.6 细胞摄取成像实验(激光共聚焦成像)
为了确定PEG-PDPA-TPP + DOX胶束的细胞摄取情况,且由于TPP自带荧光成像的作用,采用CLSM以评估细胞内胶束的摄取和分布情况。用具有相同TPP浓度的游离TPP和PEG-PDPA-TPP +DOX胶束(ρTPP= 1.0 μg/mL)分别处理Hela细胞4 h和24 h,然后加入Hoechst33342用于细胞核的染色(蓝色荧光)。如图9所示,几乎所有的卟啉荧光都是在细胞质或非核的细胞器中,这可能是因为卟啉和PEG-PDPA-TPP + DOX胶束都不具有细胞核靶向,无法穿越细胞核膜中的核孔复合物(NPS),这也与文献中的报道相符合[16,29]。随着时间的增加,无论是游离的TPP或者PEG-PDPA-TPP + DOX胶束,24 h时的TPP荧光强度都要明显强于4 h的TPP荧光强度。这表明样品的摄取是时间决定性的,随着时间的延长,细胞会摄取更多的样品,这与我们选择加药培育24 h再进行光照治疗的选择相符合。并且在4 h和24 h时,PEG-PDPA-TPP + DOX胶束(图9(b),9(d))与游离TPP处理的细胞(图9(a),9(c))对比度更强,经PEG-PDPA-TPP + DOX胶束处理的细胞所显示的TPP的荧光强度显著强于游离TPP所处理的细胞,表明相对于游离的TPP,细胞会摄取更多的PEG-PDPA-TPP + DOX胶束。这是由于游离的TPP是通过自由扩散进入细胞,而PEG-PDPA-TPP +DOX纳米粒子是通过內吞作用进入细胞,更容易被细胞摄取。

图 9 加入不同样品后的激光共聚焦成像照片(ρTPP = 1.0 μg/mL)Fig. 9 CLSM images of Hela cells incubated with different samples (ρTPP = 1.0 μg/mL)
3 结 论
(1)成功构建了在中间连接点具有TPP单元的pH响应性的两亲性嵌段共聚物,其在水溶液中的自组装可以得到TPP规则排列在核周围的球形胶束。
(2)相比于物理包裹TPP,中间连接点具有TPP单元的两亲性嵌段共聚物不仅具有确定的分子结构并能够减少卟啉的聚集,显著增强TPP的光动力效果,同时也可作为载药平台用于药物的递送。
(3)PEG-PDPA-TPP 包载 DOX胶束的pH响应释放行为可以通过肿瘤细胞摄取期间的弱酸性细胞微环境来调控。同时CLSM显示,相比于游离的TPP,将TPP连接在两亲性嵌段共聚物的组装体可以更好地被细胞所摄取。这种胶束递送系统是一种具有广泛应用前景的环境响应性和光动力与化疗协同治疗的纳米平台。
猜你喜欢
广西大学学报(自然科学版)(2022年4期)2022-09-19
实用手外科杂志(2022年2期)2022-08-31
军事文摘(2022年12期)2022-07-13
东华大学学报(自然科学版)(2022年3期)2022-06-25
少儿科技(2022年2期)2022-03-05
科学导报·学术(2020年29期)2020-10-21
商品与质量(2020年18期)2020-07-27
美与时代·美术学刊(2019年9期)2019-11-29
燕山大学学报(2015年4期)2015-12-25
外语教学理论与实践(2014年2期)2014-06-21
