
Novel frameshift mutation in the SACS gene causing spastic ataxia of charlevoix-saguenay in a consanguineous family from the Arabian Peninsula: A case report and review of literature
2020-05-14 01:51
World Journal of Clinical Cases 2020年8期
Abdullah Al-Ajmi, Neurology Unit, Al-Jahra Hospital, Jahra City 13110, Kuwait
Sarah Shamsah, Faculty of Allied Health Sciences, Kuwait University, Kuwait City 13110,Kuwait
Aleksandar Janicijevic, Department of Radiology, Al-Jahra Hospital, Jahra City 13110, Kuwait
Michayla Williams, Fahd Al-Mulla, Department of Genetics, Genatak Center for Genomic Medicine, Kuwait City 12000, Kuwait
Abstract
BACKGROUND
Familial cases of autosomal recessive spastic ataxia of charlevoix-saguenay have not been reported in the Arabian Peninsula, although the consanguineous marriage rate is very high. We report the first family from the Arabian Peninsula harboring a novel frameshift mutation in the SACS gene.
CASE SUMMARY
A 33-year-old man presented to our neurology clinic with balance problems and weakness of distal upper and lower limbs. He was previously clinically diagnosed with Friedreich's ataxia. However, the severity of polyneuropathy and the electrodiagnostic studies (EDX) findings are atypical features of Friedreich’s ataxia, and the deterioration was attributed to diabetic neuropathy. Close examination of other family members identified cerebellar ataxia, lower-limb pyramidal signs, peripheral neuropathy, and magnetic resonance imaging findings characterized by pontine linear hypointensities. Genetic testing for Friedreich’s ataxia did not yield a diagnosis. Whole exome sequencing identified a novel frameshift germline mutation in the SACS gene termed c.5824_5827delTACT using the transcript NM_014363.5, which is predicted to cause premature termination of the sacsin protein at amino acid position 1942(p.Tyr1942Metfs*9) and disrupts the sacsin SRR3 and domains downstream from it. The mutation segregated with the disease in the family.
CONCLUSION
Our data add to the spectrum of mutations in the SACS gene and argues for a need to implement suitably integrated clinical and diagnostic services, including next generation sequencing technology, to better classify ataxia in this area of the world.
Key words: Ataxia; Autosomal recessive spastic ataxia of charlevoix-saguenay; Sacsin;Autosomal recessive spastic ataxia of charlevoix-saguenay; SACS mutation; Arabia; Next generation sequencing; Case report
INTRODUCTION
Autosomal recessive spastic ataxia of charlevoix-saguenay (ARSACS-OMIM 270550)is an autosomal recessive neurological disorder that was first described in descendants of French-Perche immigrants who settled in the Charlevoix-Saguenay river region of Quebec, Canada between 1608-1760[1]. The disease was seen clustered in two isolated regions of northeastern Quebec in the period 1941 to 1985, namely Charlevoix and Saquenay-Lac-St-Jean and was clinically seen as distinct from Friedreich’s ataxia[2,3], with an estimated incidence at birth of 1/1932 and a carrier rate of 1/22 in inhabitants of Saquenay-Lac-St-Jean[4].
The ARSACS causative gene defects have been localized to chromosome 13q11[5]and were later identified as sacsin coding protein with an open-reading-frame of 11487 bp encoded by a single large exon spanning 12794 bp[6]. The sacsin multidomain protein is 520 kDa with an N-terminal Ubiquitin-like domain, 3 sacsin repeating region (SRR) domains, a J-domain (DnaJ motif of heat shock protein 40[7]), an xeroderma pigmentosum complementation group C-binding domain[8]and a C-terminal higher eukaryote and prokaryote nucleotide-binding domain[9].
ARSACS is caused by homozygous or compound heterozygous mutations in theSACSgene. In Quebec, p.Pro2948fs (frameshift) and p.Arg2502* (nonsense) represent the two major mutations in the sacsin protein[6]. The fact that unique causalSACSgene mutations were identified in Quebec accounting for 95% of the ARSACS disease alleles that were in linkage disequilibrium with other marker loci (linked to a major ancestral ARSACS haplotype) indicated a predominant mutation as a consequence of founder effect. It is now clear that pathogenicSACSgene mutations causing ARSACS are more heterogeneous and spread widely along the largeSACSgene[10]. Notably,other familial cases were reported from Tunisia[11-13], Italy[14], Japan[15], China[16,17],Brazil[18,19], Poland[20], Turkey[21,22], Holland[23], India[24], Norway[25]etc. Today, the disease is reported in at least 20 countries with more than 171 distinctSACSgene mutations published worldwide[26,27], a small number given the large size of the gene, and most likely reflecting its functional importance.
The clinical presentation of ARSACS is exemplified by the presence of three cardinal neurological signs: Firstly, early-onset and progressive cerebellar ataxia,secondly, lower-limb pyramidal signs, and thirdly, peripheral neuropathy. Many other distinct neurological disorders share the triad signs, which makes the clinical diagnosis of ARSACS difficult and necessitates a rigorous laboratory setup that benefits from periodic review of cases with the clinical team[28].
Here, we report the first family from Kuwait with ARSACS harboring a novel homozygous frameshift mutation in theSACSgene. Our study adds to the spectrum of mutations in theSACSgene and argues for a need to implement appropriate clinical and diagnostic services for the classification of ataxia in this area of the world.
CASE PRESENTATION
Chief complaints
The proband was a 33-year-old man who presented to our neurology clinic in 2013 with balance problems and weakness of distal upper and lower limbs.
History of present illness
The balance problems started in his early teens. His weakness appeared recently. His medical history was significant for diabetes mellitus.
History of past illness
The patient was diagnosed with diabetes mellitus type 2. He later developed retinal hemorrhage and a transient ischemic attack. He had poor glycemic control.
Personal and family history
The proband has two nieces and a nephew (siblings and children of his half-brother)with balance problems (Figure 1). Their ages were 18, 16, and 9 years, respectively.They all have cerebellar ataxia. In addition, the nieces have upper motor neuron signs in the lower limbs, mild distal lower limb weakness, reduced light touch, pinprick and temperature in stocking distribution, and high arch feet and hammertoes. His nieces’ nerve conduction studies showed features of demyelination and axonal loss.The nephew, also, has upper motor neuron signs in the lower limbs, mild weakness of hip flexors and ankle dorsiflexors but normal sensory examination.
Physical examination upon admission
On examination, the patient manifested typical ataxia, with mild distal weakness(more evident in the lower limbs), mild dysarthria, nystagmus, truncal and appendicular ataxia, upper motor neuron signs in the lower limbs and reduced sensation (temperature, pinprick, proprioception, and vibration in stocking distribution). He had high arch feet, and hammer-toes His general physical examination was unremarkable. His EDX showed evidence of demyelination as well as axonal loss (Tables 1 and 2).
The proband was initially diagnosed with Friedreich's ataxia. However, the severity of polyneuropathy and the EDX findings are atypical features of Friedreich’s ataxia. His workup for acquired cerebellar disorders, vitamin E levels, and cerebrospinal fluid parameters were negative/normal. He lost follow up with our clinic until March 2017. His condition deteriorated significantly. He developed muscle wasting distally in upper and lower limbs, absent DTRs throughout, loss of sensory modalities in stocking and glove distribution, in addition to cerebellar signs. He required the assistance of a wheelchair most of the time. His nerve conduction studies showed absent responses throughout
The proband score on scale for the assessment and rating of ataxia was 24. His score on the spastic paraplegia rating scale was 33.
Laboratory examinations
DNA extraction:After genetic counseling, and with informed and written consent from the family for genetic testing, peripheral blood was obtained from the proband,his parents and affected nieces and a nephew. DNA was extracted from the EDTA peripheral blood samples using DNAeasy blood and tissue kit (Qiagen, Hilden Germany) and stored until use at 4 °C.
Exome sequencing:Each sequenced sample was prepared according to the Illumina protocols and as described previously[29]. Briefly, one microgram of genomic DNA was fragmented by nebulization, the fragmented DNA was repaired, an “A” ligated to the 3′ end, Illumina adapters ligated to the fragments, and the sample was size selected. The size selected product was PCR amplified, and the final product was validated on the Agilent Bioanalyzer. Before the first hybridization, the multiple libraries were combined with different indices into a single pool prior to enrichment.The pooled DNA libraries were mixed with capture probes of targeted regions. The streptavidin beads were used to capture probes containing the targeted regions of interest. Three wash steps then removed the non-specific binding from the beads. The enriched library was then eluted from the beads and prepared for a second hybridization and sequencing on HiSeq2500 (Illumina, United States).

Figure 1 Family pedigree. Autosomal recessive spastic ataxia of charlevoix-saguenay affected members of the family are depicted with black target circle. Squares represent males and circles females. The proband is marked with an arrowhead. Notice the extensive consanguinity. Wt: Wild type; Mut: Mutant.
Read mapping:Paired-end sequences produced were first mapped to the human genome, UCSC assembly hg19 (NCBI build 37.1), without unordered sequences and alternate haplotypes, using the mapping program “BWA” (version 0.5.9rc1), and a mapping result file in SAM format using “BWA sample” was generated.
Then, the package in Picard-tools (ver.1.59) was used in order to convert the previous SAM file into a form with reads sorted by mapping-coordinate: This required the use of SortSam.jar, with the removal of PCR duplicates, thus reducing those reads identically matched to a position at the start into a single read, using MarkDuplicates.jar, which requires reads to be sorted.
Then another SAM file, including only reads that uniquely mapped to the reference genome, was created, transforming it into BAM file with the use of Samtools(ver.0.1.18). Any reads not across the targeted exonic regions were filtered out, the information of which was obtained from the manufacturer of SureSelect enrichment toolkit. This filtering is executed with the program named BED tools (version 2.15.0).
Statistics regarding those reads, such as the number of reads, its ratio to all sequences reads, and throughput was obtained from GATK (version 1.4.11).
Sanger sequencing:TheSACSgene mutation identified by exome sequence was further verified and segregated using Sanger sequencing. Genomic DNA was extracted from the patients and their family members using a standard procedure and amplified by PCR using gene-specific primers (the forward primer 5-’GAAACCACACATTGGAGAGG-3’ and the reverse primer 5’-GCTGCTGAACCAACATCTCT-3’). Genomic DNA (25 ng) was amplified at a final volume of 25 μL using GoTaq Green Master Mix-2X (Promega, United States) and 5 µmol/L primers. The reactions were performed using a Veriti®Thermal Cycler (Applied Biosystems, United States),under the following conditions: initial denaturation cycle at 95 °C for 5 min; followed by 35 extension cycles of 95 °C for 45 s, 58 °C for 45 s and 72 °C for 1 min; a final extension cycle at 72 °C for 7 min, and amplicons were kept at 4 °C. Then, the amplified PCR products were purified using ExoSAP-IT (Affymetrix, United States)and sequenced using Big Dye Terminator cycle sequencing kit (Applied Biosystems,United States) as described by the manufacturers. Bi-directional sequencing reactions were carried out on each purified product. A DyeEx 2.0 Spin Kit (Qiagen, Germany)was used to remove unincorporated dye within the sequencing products, which were loaded on the ABI Prism 3730xl Genetic Analyzer (Applied Biosystems, United States)for sequencing. The resulting sequence contigs were analyzed and aligned against a reference sequence using the Chromas Pro software to detect the sequence variations.
Depersonalized data from the proband is made available on our web server at the following address: https://www.genatak.org/public-data.
The excel sheet contains all the variants from the proband identified by exome sequencing. Moreover, the full sequences will be submitted to Genesis 2.0 in due course.
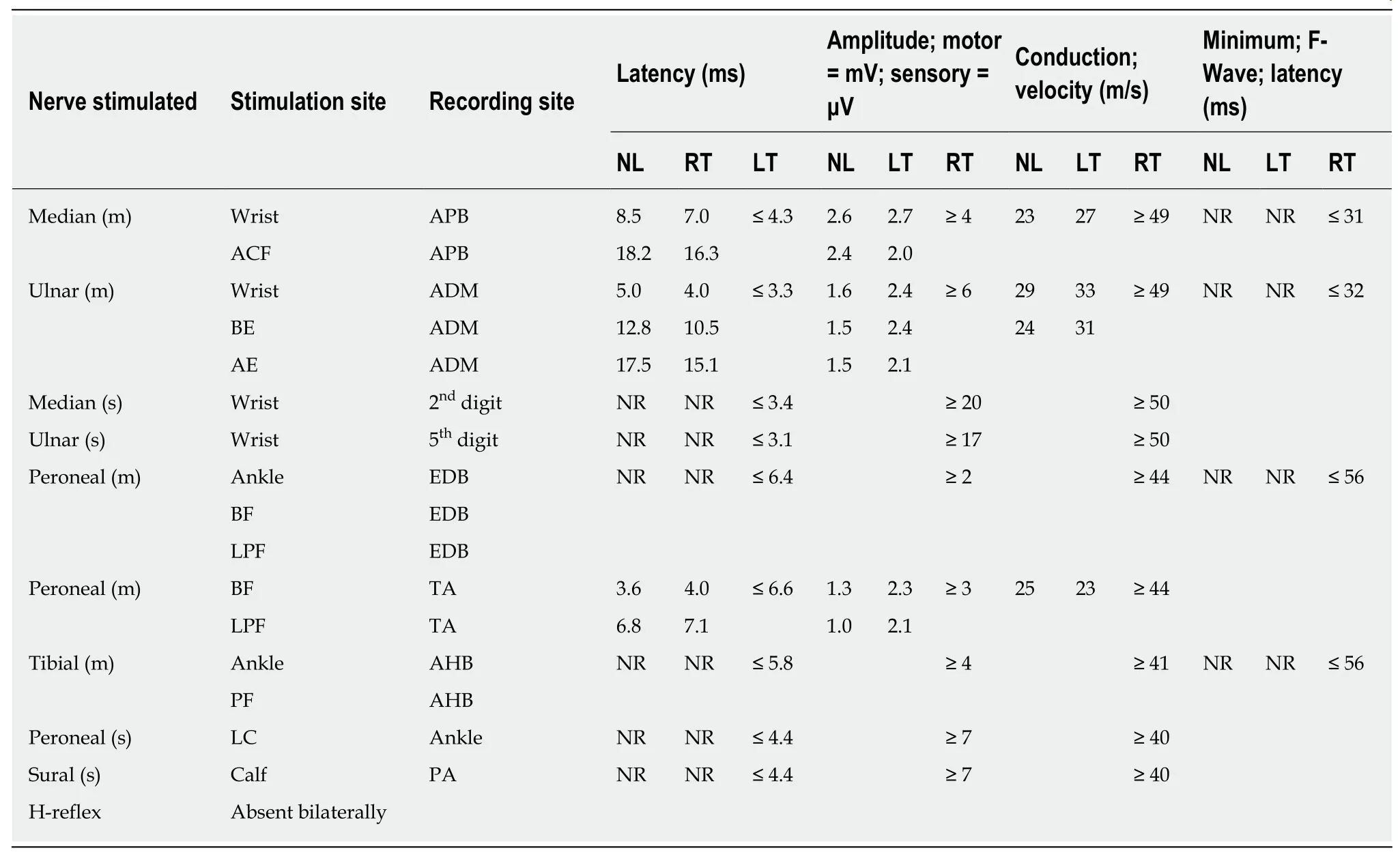
Table 1 Nerve conduction studies of proband II-3
If required, VCF exome-data files can also be accessed by academics after registering on the website and signing a confidentiality agreement.
Imaging examinations
The proband imaging workup showed the characteristic radiologic manifestations of ARSACS. These included profound cerebellar atrophy predominately at the superior vermis with enlargement of the supravermian cisterns and cisterna magna, and superior spinal cord atrophy seen by computed tomography and magnetic resonance imaging (MRI) imaging. Specific MRI imaging findings were T2WI/FLAIR hypointense paramedian pontine linear striations-“tigroid“ pattern, involving the upper and middle pons, not reported in other causes of ataxia or spastic paraparesis.Associated with pontine linear hypointensities may be diffuse T2WI/FLAIR hyperintense signal involving the lateral pontine aspects merging to the middle cerebellar peduncles. Other imaging findings included thinning of the posterior body of the corpus callosum, cerebral (post central and parietal) atrophy (Figure 2). The nieces' MRI brain images showed similar abnormalities (data not shown).
Genetic analysis
The patient was referred to the medical genetics services at the Genatak center for Genomic Medicine (Kuwait) where he and the extended family were counseled,assessed and they consented for exome sequencing of the proband at a depth of 150x and GAA repeat length estimation of theFDRAgene. The extensive consanguinity present in the pedigree was consistent with an autosomal recessive pattern of inheritance (Figure 1).
Genetic testing for Friedreich’s ataxia was negative. TheFRDAgene contained two GAA repeats in the normal range of 7 and 19 GAA trinucleotide repeats (data not shown).
The initial quality of exome sequencing yielded 10.49 Gigabases of data totaling 103886270 reads with Q30 of 97.10% (The quality ratio satisfying Phred quality score greater than 30, which represents an error rate of 1 in 1000, with a corresponding call accuracy of 99.9%). Before filtering, exome sequencing identified 169489 single nucleotide polymorphisms, insertions and deletions (Figure 3). We next employed a carefully designed filtering procedure that is dependent on high read depth, rarevariants calling, and other matrices shown in Figure 2 to identify 2 genes with known ARSACS association. One was an apparently homozygous frameshift deletion in exon 10 of theSACSgene (Figure 4A), termed c.5824_5827delTACT using the transcript NM_014363.5, which is predicted to cause a premature termination of the sacsin protein at amino acid position 1942 (p.Tyr1942Metfs*9) and disrupt the sacsin SRR3 and domains downstream from it (Figure 4B). The mutation is novel and has not been described in any of the public databases, including the 1000 human genome project and gnomAD databases. Next, we confirmed the homozygousSACSgene frameshift mutation in the proband (II-3) by Sanger sequencing (Figure 5). Both normal parents(I-1 and I-2) of the proband (II-3) were obligatory carriers/heterozygous for the mutation (Figure 5). We next segregated the mutation in the affected nieces (III-1 and III-2) and nephew (III-5). All were homozygous for the same mutation, and their unaffected consanguineous parents (II-6 and II-7) were heterozygous carriers (Figure 5). Functional studies will, however, be required to assess the reported mutation impact on theSACSgene function.
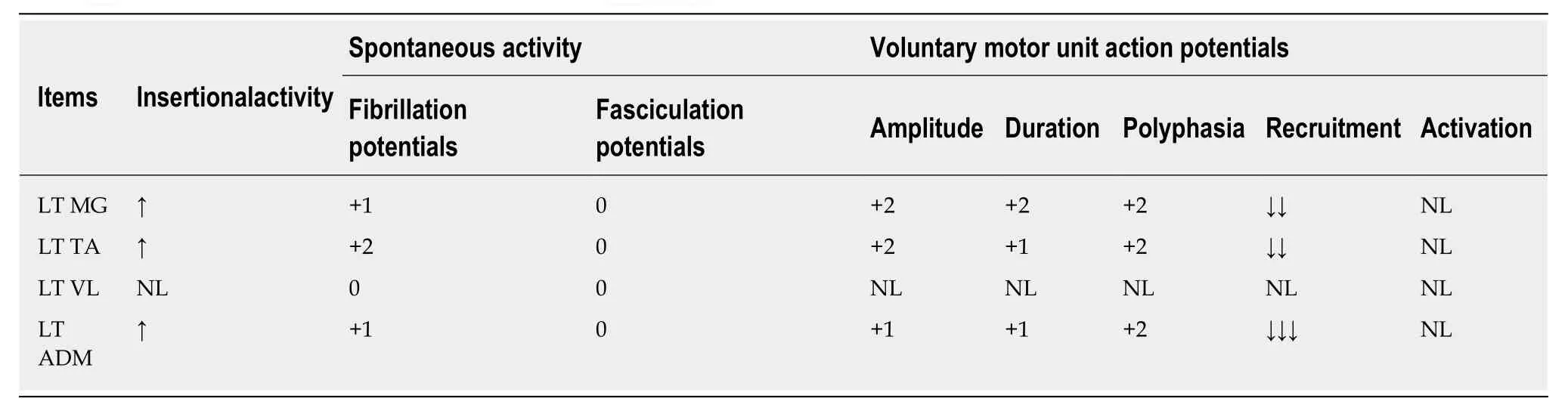
Table 2 Needle electromyography of proband II-3
FINAL DIAGNOSIS
The patient was finally diagnosed with ARSACS-OMIM 270550.
TREATMENT
The patient has been doing physiotherapy and management of his diabetes but later developed diabetic retinopathy and has been followed up with an ophthalmologist.Optical coherence tomography is not available.
OUTCOME AND FOLLOW-UP
Overall the patient deteriorated gradually. The worsening of his conditions may have been accelerated by diabetes mellitus with its complications (microvascular and polyneuropathy).
DISCUSSION
To the best of our knowledge, ARSACS has never been described in the Arabian Peninsula before this report. Given the high prevalence of consanguinity in Arab countries, which amounts to 50%-70% of marriages[30,31], the lack of reported ARSACS cases is intriguing. While selective inbreeding may have contributed to eliminating theSACSgene mutations in Arabia, it is more likely that the lack of reported cases from the Arabian Peninsula reflects a misdiagnosis of the disease. Indeed, the diagnosis of ARSACS is challenging[27,28]. The clinical presentations are closely mimicked by other neurodevelopmental disorders requiring a dedicated multidisciplinary team composed of clinical scientists, neurologists, radiologists, and geneticists to collaborate effectively. Unless such an elaborate system is strictly adhered to, ARSACS, and other complex genetic disorders, may be misdiagnosed.

Figure 2 Brain magnetic resonance imaging images of the proband II-3. A: T2WI transversal cut of the middle pons showing hypointense linear striations,conforming to the tigroid pattern; B: T2WI sagittal images showing atrophy of the cerebellum, predominantly at the superior vermis and relative thinning of the posterior body of the corpus callosum, reduced diameter of the cervical spinal cord. Prominent sulci, fissures and extra-axial spaces, advanced to the age of the patient; C: T2WI coronal cut showing diffuse hyperintense signal involving the lateral pontine aspects merging to the middle cerebellar peduncles.
The family we reported here satisfied the cardinal clinical signs and symptoms of ARSACS, namely cerebellar ataxia, lower limb pyramidal track signs, axonaldemyelinating sensory-motor peripheral neuropathy, and linear pontine T2-hypointensities described previously[2,27,32,33]. However, given the age differences of the affected members of the family, we saw time-dependent differences in the presenting phenotypes where the youngest individual (III-5) was minimally affected. This progressive neurodegeneration phenomenon may further complicate the ARSACS diagnosis. It has been shown that some pathogenicSACSgene mutations present without cerebellar atrophy or peripheral neuropathy[27], which argues for the use of next generation sequencing technology in all cases of ataxia regardless of the presence or absence of ARSACS cardinal signs. Indeed, the proband in this family was referred to the Genatak Genomic Medicine Center for exome sequencing after years of delay and failure of other genetic tests to reach a diagnosis. The pathogenicSACSgene mutation described here segregated with the disease and appeared to disrupt twothirds of the gigantic exon 10 leading to loss of SRR3 and downstream domains that are critical for sacsin ATPase function and dimerization[34].
The function of the gigantic 520 kDa multimodular sacsin protein is not well understood[34]. Functional studies have been scarce but fruitful. Girardet al[35],localized sacsin protein to the mitochondria and have shown a relationship between loss of sacsin function and reduced mitochondrial fission.
By documenting the first family in the Arabian Peninsula, we argue that ARSACS is likely to be underdiagnosed. The extensive use of next generation sequencing,therefore, may illuminate further families in this area with the disease given the high rate of consanguineous marriages. Policymakers and Local health providers should be encouraged to expand the utilization of next-generation sequencing in screening programs and prenatal diagnosis, which will ultimately improve the diagnosis and treatment of complicated neurological disorders with an obvious positive impact on genetic counseling competences.
CONCLUSION
We have identified a novelSACSgene frameshift mutation in a consanguineous family from the Arabian Peninsula diagnosed for the first time with ARSACS. We hope that this data will encourage health providers to adopt our multidisciplinary approach of care and implement next generation sequencing as the primary technology in the investigation of ataxia.

Figure 3 Flowchart representing the filtering strategy employed in the analysis of exome sequencing to reach a diagnosis in the proband II-3. UTR:Untranslated region.


Figure 4 SACS gene location and N- to C-terminal of the sacsin protein. A: Exome sequencing summary layout depicting the SACS gene location on chromosome 13 at the top and the exons coverage in green and blue. The location of the mutation on exon 10 is represented by a vertical black line. The deletion TACT mutation in the zoomed view is shown in magenta, and the reference sequence marked with a rectangle; B: N- to C-terminal of the sacsin protein, depicting a ubiquitin-like domain that binds to the proteosome, three sacsin repeat regions with known Hsp90-like chaperone function, an xeroderma pigmentosum complementation group C-binding domain that binds to the Ube3A ubiquitin protein ligase, a DnaJ domain that binds Hsp70, and a higher eukaryotes and prokaryotes nucleotide-binding domain that facilitates sacsin protein dimerization. The horizontal black l line depicts the start of the mutation, which causes loss of function of the protein due to the loss of all amino acids downstream of the line. The blue arrow shows the disrupted protein sequences and domain of sacsin protein. Each domain is demarcated by a specific sequence highlighted by the corresponding color and amino acid number given that methionine is the first amino acid at the N-terminus.
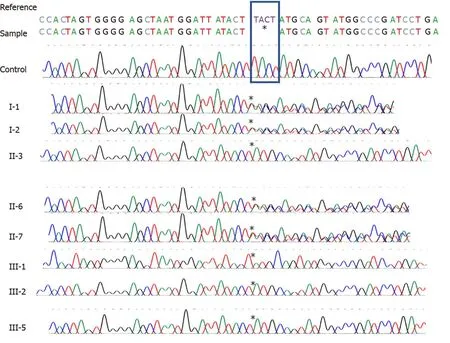
Figure 5 Sanger sequencing of control individual and the family in Figure 1A, including the proband and his parents. The rectangle depicts the 4 deleted bases in the SACS gene. The asterisks mark the base before and the base after the deletion.
 World Journal of Clinical Cases2020年8期
World Journal of Clinical Cases2020年8期
- World Journal of Clinical Cases的其它文章
- Probiotic mixture VSL#3: An overview of basic and clinical studies in chronic diseases
- Mucosa-associated lymphoid tissue lymphoma simulating Crohn’s disease: A case report
- Embolization of pancreatic arteriovenous malformation: A case report
- Duodenal mature teratoma causing partial intestinal obstruction: A first case report in an adult
- Rare anaplastic sarcoma of the kidney: A case report
- Unusual association of Axenfeld-Rieger syndrome and wandering spleen: A case report