
环己烯环氧化反应催化剂研究进展
2020-08-07 09:54王新国程庆彦彭文静王明明王延吉
高校化学工程学报 2020年3期
王新国, 程庆彦,2, 彭文静, 王明明, 王延吉,2
(1. 河北工业大学 化工学院, 天津 300130; 2. 河北省绿色化工与高效节能重点实验室, 天津300130)
1 引 言
环氧环己烷作为一种重要的有机中间体,由于具有活泼的环氧基,因此很容易与胺、酚、醇、羧酸等物质反应,生成附加值更高的化合物[1-3]。随着苯选择性加氢制备环己烯工艺的成熟,催化环己烯环氧化制备环氧环己烷的工艺倍受关注。环己烯具有双键和烯丙基两个氧化活性位点,双键环氧化生成环氧环己烷,环氧环己烷进一步水解生成1,2-环己二醇,1,2-环己二醇被氧化开环生成己二酸[4-5]。烯丙基α-H受到双键的影响,C-H 键能相对较低,发生烯丙基氧化生成2-环己烯-1-醇和2-环己烯-1-酮,烯醇烯酮进一步环氧化生成7-氧杂二环[4.1.0]庚烷-2-酮(图1)[6]。环己烯环氧化制备环氧环己烷伴随着过度氧化以及烯丙基氧化副反应,因此利用环己烯环氧化制备环氧环己烷面临着巨大的挑战。本文对环己烯环氧化反应体系中催化剂种类进行分析,希望能对学术研究以及工业生产提供一些帮助。

图1 环己烯氧化路线[6]Fig.1 Reaction paths of cyclohexene oxidation[6]
2 氧气作为氧化剂的催化体系
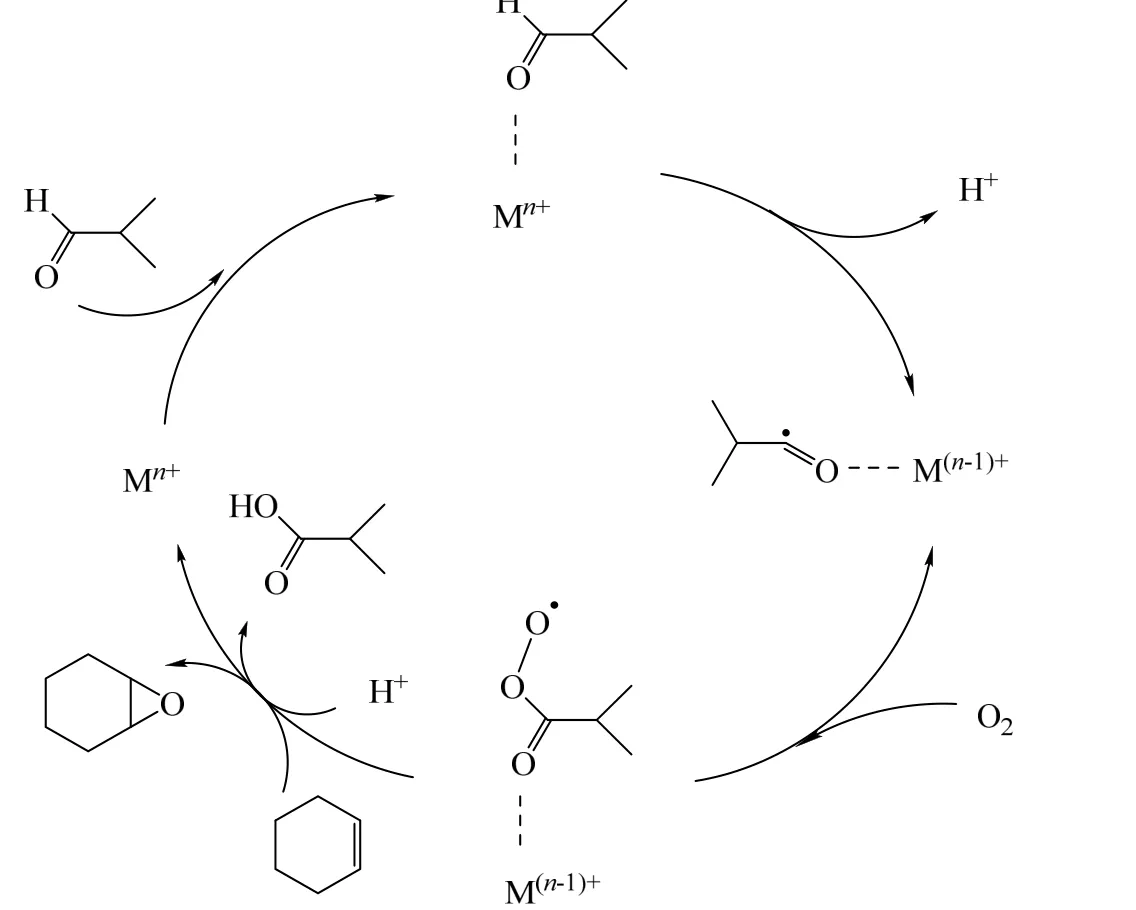
图2 异丁醛助氧化剂存在下氧气氧化环己烯环氧化的反应机理[16]Fig.2 Epoxidation mechanism of cyclohexene using molecular oxygen as the oxidant and isobutyraldehyde as the co-oxidant[16]
氧气由于价廉易得、原子经济性高等特点,已成为一种潜在的理想氧化剂,被广泛用于氧化醇、醛、烷烃、烯烃等物质[7-10]。使用氧气作为氧化剂氧化环己烯,选择性一般较低,很容易造成过度氧化生成2-羟基环己酮、7-氧杂二环[4.1.0]庚烷-2-酮、己二酸等副产物,国内外学者做了大量研究,致力于以分子氧作为氧化剂氧化环己烯制备环氧环己烷[6,11-12]。
20 世纪 90 年代,YAMADA 等[13]首先发现二乙酰丙酮镍配合物在异丁醛存在下可催化环己烯环氧化反应,醛的存在不仅能提高催化活性还能提高产物选择性(表1,entry 1)[14-15]。在异丁醛存在下氧气氧化环己烯环氧化的反应机理(图2)中[16],醛的C-H 键被活性中心活化生成醛基自由基,然后结合氧分子从而完成氧的转移,因此能够引发自由基反应的过渡金属Co、Mn、Cu、Fe 等配合物被研究报道[17-25]。
钴类催化剂在环己烯环氧化反应中具有较高的催化活性被广泛研究,在异丁醛存在下,SUN 等[17]用酞菁四羧酸Co 配合物(Co-PTC)催化环己烯环氧化,环己烯转化率100%,环氧环己烷的选择性58.1%(表 1,entry 2)。SALAVATI-NIASARI 等[18]将席夫碱 Co 配合物负载在多壁碳纳米管上(Co((OH)2-salophen)/MWNTs)用于催化环己烯环氧化,环氧环己烷收率可达88%,催化剂重复使用3 次,转化率和选择性几乎没有下降(表1,entry 3)。阳离子Co 卟啉配合物(CoTMPyP)通过离子交换技术负载在层状化合物K4Nb6O17上用于环己烯环氧化,负载后的催化剂相对于卟啉钴催化剂,环氧环己烷选择性提高了 13.4%;催化剂重复使用 5 次,环己烯转化率略有下降后趋于平稳,而环氧环己烷选择性从94.3% 增加到98.1% (表1,entry 4)[19]。由于过渡金属配合物催化的环己烯烯丙基氧化也属于自由基反应,因此具有较高催化活性的Co 配合物及其负载催化剂很难避免2-环己烯-1-醇和2-环己烯-1-酮的生成[20]。
活性较弱的Mn 相对于Co 配合物催化环己烯环氧化,表现出更优秀环氧环己烷选择性。ZHAO 等[21]将丙氨酸席夫碱锰配合物(Sal-Ala-Mn)锚定在二氧化硅上用于环己烯环氧化,环己烯的转化率达到 99.6%,环氧环己烷选择性88.2% (表1,entry 5)。卟啉锰配合物 (MnP) 固定在苯乙烯-羟乙基甲基丙烯酸酯共聚物上(P(St-co-HEMA)MnP)催化环己烯环氧化,当异丁醛:环己烯 = 3:1(摩尔比)时,环己烯转化率 > 99%,环氧环己烷选择性93.0% (表1,entry 6)[22]。采用Mn 卟啉配合物(Mn(TPP)Cl)作为催化剂,在牺牲异丁醛利用效率的前提下,采用5 倍摩尔量的异丁醛作还原剂,环己烯转化率 > 99%,环氧环己烷选择性 >99% (表1,entry 7)[23]。随着异丁醛的用量不断增大,环氧环己烷的选择性得到提升,但是当环己烯:异丁醛= 1:1(摩尔比)时,环氧环己烷的选择性只有90% 左右,同时伴随着更多的烯丙基氧化产物生成[24]。异丁醛的加入必然产生异丁酸,因此在保证高选择性的同时,如何提高异丁醛的利用效率便成为首要问题。
Cu、Ti、Fe 等过渡金属 (表1,entry 8~11)[25-28]也被用于环己烯氧化,其中PMo11Fe/SBA 催化环己烯环氧化的转化率仅为18%,环氧环己烷的选择性为99%,虽然环己烯的转化率较低,但是超高环氧环己烷的选择性引人注目(表1,entry 9)[26]。
不加入异丁醛高选择性的有氧氧化环己烯制备环氧环己烷虽然符合绿色化学的要求,但是很容易发生烯丙基氧化, 目前仅有少量文献报导。 RAO 等[29]合成席夫碱氧化钼配合物(cis-[MoO2(hap-SMDTC)(solv)])用于环己烯环氧化,氧气作为氧化剂,环己烯的转化率 87%,环氧环己烷的选择性85% (表1,entry12)。SHRINGARPURE 等[30]将磷钨酸根负载在三氧化二铝上(PW11/Al2O3)用于环己烯环氧化,无溶剂条件下环己烯的转化率85%,环氧环己烷的选择性100%,催化剂重复使用3 次,选择性没有下降(表1,entry13)。

表1 氧气氧化环己烯环氧化Table 1 Epoxidation of cyclohexene by O2
从分子轨道理论分析,基态的分子氧为三线态,基态的环己烯分子为单线态,分子氧与有机分子的作用是自旋禁阻的,因此二者不能反应[31]。过渡金属对分子氧的活化能够打破自旋禁阻,使反应顺利进行。当氧分子被过渡金属活化后仍然很难发生氧原子的转移而完成环氧化反应,更多的是发生烯丙基C-H 氧化[32]。当加入异丁醛后,氧气优先与异丁醛反应生成醛基过氧化物,真正发挥氧原子转移的化合物是异丁醛过氧化物[16],由于醛基的C-H 键能比烯丙基C-H 键能更低,因此反应控制在较低的温度下进行 (表 1),较低的反应温度能够活化氧气氧化醛基C-H 键,同时避免烯丙基C-H 键的氧化,从而减少了副产物环己烯醇和环己烯酮的生成。活性较弱的Mn 类催化剂,也是由于避免了烯丙基C-H 键的氧化而提高了产物选择性。
3 过氧化氢作为氧化剂的催化体系
过氧化氢相对于氧分子拥有更高的氧化选择性,因此广泛用于氧化有机化学品制备附加值更高的醇、醛、酮、环氧化合物等[33-34]。作为一种绿色的氧化剂氧化环己烯,理论上副产物只有水生成,因此更符合绿色化学的理念。含有过渡金属 Ti,W、Mn 等催化剂被广泛用于催化双氧水环氧化环己烯,取得了较好的催化效果[35-48]。
CHEN 等[35]采用TiO2催化双氧水氧化环己烯,环己烯转化率为42%,环氧环己烷选择性为54.8%,值得注意的是,吡啶红外分析结果发现TiO2表面存在大量L 酸和B 酸酸性中心(表2,entry 1)。L 酸和B酸催化环氧环己烷开环水解[36],以及 TiO2活化双氧水氧化环己烯发生烯丙基氧化[37],因此 TiO2催化环己烯环氧化的选择性相对较低。钛硅分子筛(TS-1、Ti-MWW、Ti-MCM-41)作为一种重要的催化剂被广泛用于催化环己烯环氧化研究,其中TS-1 催化环己烯环氧化具有良好的产物择形性,但是催化环己烯环氧化的转化率通常只有1%~4% (表2,entry 2)[38]。VERNIMMEN 等[39]将TS-1 分散在SBA-15 介孔二氧化硅上 (TS-1/SiO2-pH13),环己烯转化率为6%,环己烯在微孔分子筛中的内扩散是影响催化活性的主要因素 (表2,entry 3)。为了降低内扩散阻力,分级孔TS-1 分子筛被应用于催化环己烯环氧化。JIAO 等[40]用四丁基氢氧化铵 (TPAOH)处理 TS-1 制备分级孔钛硅分子筛 (h-TS-1-T),使用有机碱腐蚀分子筛骨架以制备大孔径的通道,环己烯的转化率提升到19%,但是环氧环己烷的选择性略有降低,生成更多的1,2-环己二醇 (表2,entry 4)。而以丙醇和TPAOH 处理TS-1 分子筛 (h-TS-1-TP),环己烯的转化率提高到36.1%,同时获得96%的环氧环己烷选择性 (表2,entry 5)[41]。孔径略大于TS-1 的Ti-MWW 分子筛催化环己烯环氧化活性更高,当H2O2:环己烯 = 1(摩尔比)时,环己烯的转化率达到35%,环氧环己烷的选择性达到93%~95% (表2,entry 6)[42]。当孔径继续提升达到介孔时,Ti-MCM-41 催化环己烯氧化,H2O2:环己烯= 2 (摩尔比)时,环己烯的转化率达到56%,同时环氧环己烷的选择性几乎没有降低(表2,entry 7)[39]。随着分子筛孔径的变大,催化剂的催化活性明显增强,介孔钛硅分子筛由于结构的完整性相对于碱处理的TS-1拥有更高的产物选择性。
钨类催化剂具有催化环己烯环氧化性能,在酸性条件下,Na2WO4催化环己烯环氧化并进一步氧化开环制备己二酸,甚至可以达到100% 的己二酸收率[43]。在中性或弱碱性条件下,虽然Na2WO4和WO3催化环己烯环氧化的选择性接近100%,但是环己烯的转化率仅有0.5%和2.9% (表2,entry 8)[44]。为提高催化剂的催化活性,钨硅分子筛、有机化合物修饰的氧化钨成为研究的热点。MAHESWARI 等[44]合成分子筛W-SBA-16 催化剂,当Si:W = 20 (摩尔比)时,环己烯的转化率最大可以达到22%,虽然催化活性明显增强,但是烯丙基产物2-环己烯-1-醇明显增多 (表2,entry 9)。WU 等[45]利用烷烃咪唑基阳离子修饰的过氧化钨 ([D12mim]1.5PW4O24) 催化双氧水氧化环己烯,环氧环己烷的收率达到 94%,更有趣的是双氧水的用量不足,但环己烯转化率和选择性都很高 (表2,entry 10)。MOURET 等[46]以十二烷基三甲基溴化铵修饰的磷钨酸根(C12-PW12O40)催化环己烯环氧化,在环戊基甲基醚或 2-甲基四氢呋喃溶剂中,环己烯转化率均大于95%,同时C12-PW12O40对H2O2的利用效率也较高,在环己烯:H2O2= 1:1 (摩尔比)时,双氧水几乎全部进行环氧化反应 (表2,entry 11)。
过渡金属 Mn 配合物具有催化环己烯环氧化性能,聚乙烯亚胺 Mn 配合物固载在四氧化三铁上(Fe3O4@PEI.Mn),以碳酸氢钠为助剂,环己烯的转化率由3% 提高到48%,当H2O2:环己烯 = 4:1 (摩尔比)时,副产物只有2-环己烯-1-醇,随着过氧化氢用量的增多,2-环己烯-1-醇、2-环己烯-1-酮成为主要的产物 (表2,entry 12)[47]。由于Mn 配合物既有催化环己烯环氧化性能,又有催化环己烯烯丙基氧化的特性,因此Mn 配合物催化环己烯环氧化很难避免烯丙基副产物的生成[48]。适当减少双氧水的用量,虽然降低了环己烯的转化率,但是副产物2-环己烯-1-醇、2-环己烯-1-酮的选择性明显降低(表2,entry 13)[49]。
其他金属配合物也被用于催化环己烯环氧化,THORNBURG 等[50]将4-叔丁基-杯[4]芳烃Ta 配合物负载在SiO2上 (Calix-Ta-SiO2) 催化环己烯环氧化,环氧环己烷和1,2-环己二醇的总选择性达到了98%,而Ta 负载在 SiO2上,2-环己烯-1-醇和 2-环己烯-1-酮副产物选择性提高到 20% ~30% (表 2,entry 14)。更有趣的是,非血红Mn 化合物([MnIV(O)(N4Py)]2+)催化环己烯氧化,当环己烯为原料时,主要生成2-环己烯-1-醇,当氘代环己烯作原料时,主要生成环氧环己烷(图3),由此可见,环己烯烯丙基氧化与双键环氧化的活化能较为接近[51]。由于金属配合物具有环氧化和烯丙基氧化双重催化性能,因此催化环己烯环氧化很难避免烯丙基氧化产物的生成,且双氧水的有效利用效率也较低[52]。非金属催化剂由于催化机理的改变,反应条件更加温和,TYABLIKOV 等[53]将 2.2.2-三氟苯乙酮固定在 SBA-15 介孔二氧化硅上(F-C6H4-SBA)用于环己烯环氧化,在25 ℃下,环己烯的转化率30%,环氧环己烷的选择性100% (表2,entry 15)。

图3 环己烯和氘代环己烯的氧化路线以及收率Fig.3 Oxidation reaction paths and yield of cyclohexene and cyclohexene-D10
过氧化氢是一种清洁、高氧化选择性的氧化剂,多数氧化反应采用过氧化氢水溶液,催化剂上的 B酸L 酸会活化水分子,并与环氧环己烷反应生成 1, 2-环己二醇。为避免环氧环己烷的水解,催化剂不应具有B 酸或 L 酸酸性位点[34]。相对于异丁醛氧气体系 (表 1),H2O2作为氧化剂时反应温度较高 (表2),这主要是由于,在不添加异丁醛助剂的条件下,H2O2氧化环己烯环氧化需要更高的反应温度才能实现氧原子的转移[38]。另外,H2O2的均裂生成羟基自由基是引发烯丙基副反应的关键因素[54],为了保证反应体系不会沿自由基路线反应,反应体系的温度又不宜过高,催化剂必须没有自由基激发活性位点。钛硅分子筛以及磷钨酸根等催化剂,由于不含酸性位点以及较强的自由基激发活性位点,在催化H2O2氧化环己烯环氧化的反应中表现出较佳的催化性能[39,43]。
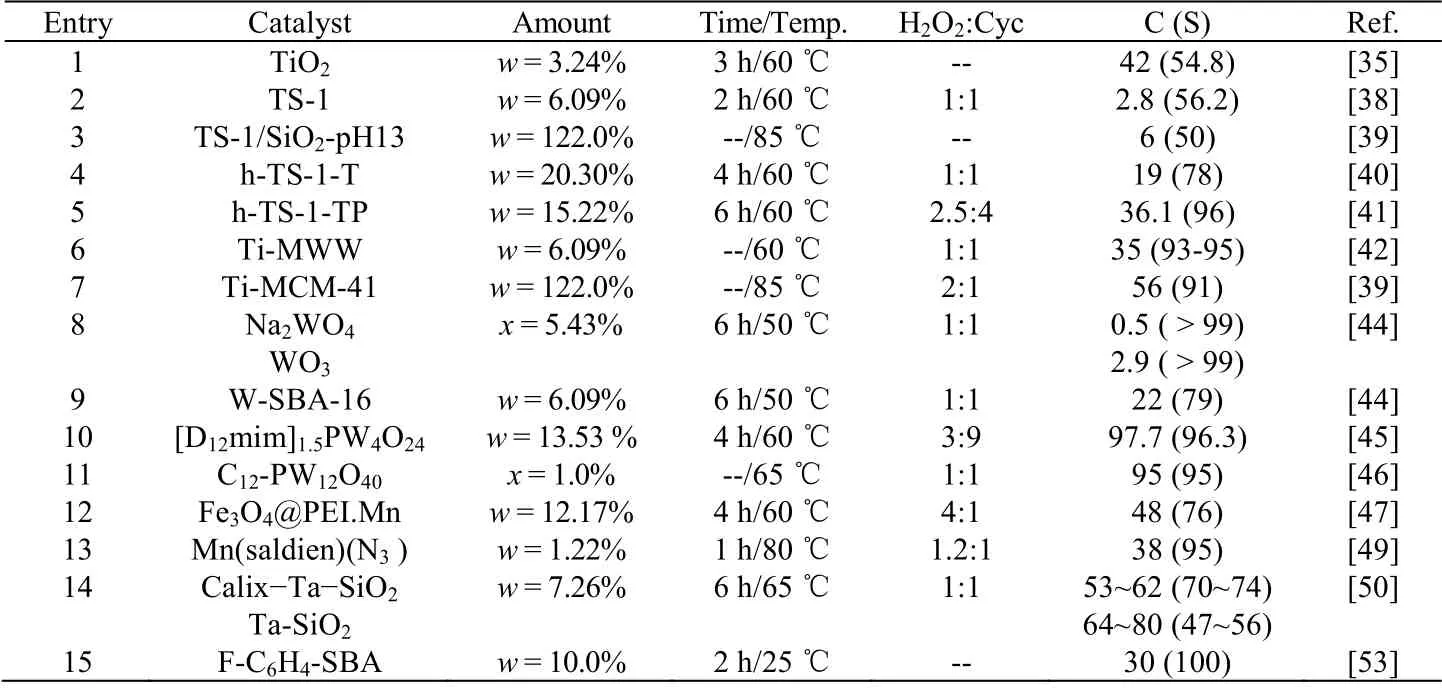
表2 过氧化氢氧化环己烯环氧化Table 2 Epoxidation of cyclohexene by H2O2
4 叔丁基过氧化氢作为氧化剂的催化体系
叔丁基过氧化氢(TBHP)作为一种烷基氢有机过氧化物比过氧化氢热稳定性更好,氧化剂的有效利用率更高[55]。相对于枯基氢过氧化物、尿素过氧化氢、亚碘酰苯等其他有机过氧化物,TBHP 还原后只产生叔丁醇,叔丁醇易于从反应体系中分离,进行循环使用[56-59]。TBHP 被V、Mo、Ti 等过渡金属活化氧化环己烯制备环氧环己烷的收率更高,因此TBHP 广泛用于氧化环己烯环氧化反应[57,60-70]。
MAURYA 等[60]合成氧化钒席夫碱配位聚合物([-CH2{VO(sal-dach)DMF}-]n)用于催化环己烯氧化,当环己烯:TBHP = 1:0.5(摩尔比)时,环己烯的转化率为51%,环氧环己烷的选择性为58%,副产物主要是烯丙基氧化产物2-环己烯-1-酮和2-环己烯-1-醇(表3,entry 1)。活性中心氧化钒兼具催化环己烯双键环氧化和烯丙基氧化的两种特性,El-KORSO 等[61]将氧化钒负载在氧化铈上(VO2/CeO2),以TBHP 作为氧化剂用于环己烯环氧化,随着钒的负载量不断增大,环己烯的转化率不断降低,环氧环己烷的选择性不断提高。当钒的含量达到 15% 时,环氧环己烷的选择性达到 90%。值得注意的是当钒的负载量进一步增加到20% 时,环氧化反应路线完全关闭,100% 的生成烯丙基类氧化物(表3,entry 2)。
钼类催化剂应用于催化 TBHP 氧化环己烯,比氧化钒催化氧化环己烯的选择性更高。仅以 MoO3作为催化剂,催化剂活性不高,环氧环己烷的选择性也较低[62]。HAMDY 等[62]合成钼硅分子筛Mo-TUD-1,以其催化环己烯氧化,环己烯的转化率提高到22% (表3,entry 3)。为使活性组分MoO3得到充分分散,各种负载型MoO3被研究报道[63-64]。BEHERA 等[63]采用浸渍法将氧化钼负载在磷酸氧钒上(Mo-VPO),环己烯的转化率提高到100% (表3,entry 4)。将氧化钼负载在ZrO2上(MoO3/ZrO2),环氧环己烷的收率甚至达到了97%,但是催化剂存在活性组分容易流失的问题[64]。而以新型材料(金属有机骨架化合物)MOFs 作为载体,完美解决了活性组分的流失,NOH 等[64]将氧化钼活性组分沉积在MOFs节点上合成Mo-NU-1000,催化环己烯氧化达到接近100% 环氧环己烷选择性,93% 环己烯转化率,催化剂使用一次仅有微量活性组分浸出 (表3,entry 5)。此外,氧化钼席夫碱配合物催化环己烯氧化,当反应体系中存在水时,环氧环己烷很容易开环水解[65]。由于席夫碱也能够水解,因此席夫碱配合物的稳定性也是催化剂能否发挥作用的前提[66]。为了防止环氧环己烷的水解以及催化剂的结构损坏,ROMANOWSKI 等[67],以二氧化钼席夫碱配合物(MoO2/L1)作为催化剂,TBHP 的癸烷溶液作为氧化剂,从而减少反应体系中水的含量,环氧环己烷的选择性提高到96%,因为水是反应产物之一,所以仍然有少量1, 2-环己二醇生成(表3,entry 6)。
Ti 配合物催化TBHP 氧化环己烯极易沿自由基反应路线生成烯丙基氧化产物[68],因此环氧环己烷选择性不高。TiO2催化TBHP 氧化环己烯转化率较低,但是将TiO2负载在SiO2上(TiO2/SiO2),环己烯转化率提高到80.6%,环氧环己烷选择性97.8% (表3,entry 7)[57]。利用硫酸铵或是碳酸铵加TPAOH 处理TS-1制造分级孔钛硅分子筛(TS-1-P-(NH4)2SO4、TS-1-P-(NH4)2CO3),环己烯的转化率为45%,环氧环己烷选择性为94% (表3,entry 7)[57]。而介孔分子筛Ti-MCM-22 由于孔径的扩大,不需要碱处理,环己烯的转化率达到41.5%,同时兼具接近100% 的环氧环己烷选择性(表3,entry 8)[69]。由于TBHP 更加稳定,有效利用率普遍大于90%[70],虽然TBHP 比H2O2价格更高,但是有效利用率完全弥补了价格缺点。其他过渡金属Fe、Cu、Co 等催化剂虽然也可以催化TBHP 氧环环己烯环氧化,但是催化环己烯烯丙基氧化特性很难避免2-环己烯-1-醇和2-环己烯-1-酮副产物的生成[71-73]。
TBHP 与H2O2较为相似,TBHP 由于热稳定性更高,不容易生成自由基而避免了烯丙基氧化反应,但是为了活化TBHP 实现氧原子的转移,反应温度进一步提高(表3)。烯丙基氧化反应不仅与氧化剂的种类有关,同时催化剂的活性中心也对产物选择性有较大的影响,催化活性较高的 Fe、Cu、Co 等催化剂催化环己烯氧化,仍然会发生自由基反应生成烯丙基氧化产物[74]。活性较弱的 Ti、Mo 类催化剂,由于催化TBHP 生成自由基的活性较弱,结果导致环氧环己烷的选择性较高[54,61]。

表3 叔丁基过氧化氢氧化环己烯环氧化Table 3 Epoxidation of cyclohexene by TBHP
5 结论与展望
环氧环己烷作为一种重要的中间体,能够进一步反应生成附加值更高的化合物,因此高效催化环己烯制备环氧环己烷具有重要的意义。本文以氧化剂进行分类,对催化剂种类以及催化体系面临的问题进行综述。分析得到催化剂的酸性位点以及自由基激发活性位点是环氧环己烷水解、烯丙基氧化两大副反应产生的主要原因。为催化剂的设计提供了方向,含有 Ti、W、Mo 等过渡金属的分子筛、氧化物表现出较佳的催化选择性。此外,催化剂的活性受催化剂表面极性的影响,极性越强的催化剂与环己烯的接触越困难,催化活性越低。为了有效提高催化活性,表面有机修饰以及有机载体的利用都是重要的研究方向。
猜你喜欢
中国调味品(2022年12期)2022-12-05
中氮肥(2022年3期)2022-05-31
科学导报(2022年28期)2022-05-24
河南化工(2021年10期)2021-11-10
化工管理(2021年7期)2021-05-13
林产化学与工业(2021年2期)2021-05-11
环境保护与循环经济(2020年4期)2020-06-08
山东化工(2020年5期)2020-04-07
中国特种设备安全(2019年1期)2019-03-13
中国塑料(2015年1期)2015-10-14
