
Structural elucidation of SARS-CoV-2 vital proteins:Computational methods reveal potential drug candidates against main protease,Nsp12 polymerase and Nsp13 helicase
2020-09-04 09:33MuhammadUsmanMirzaMatheusFroeyen
Muhammad Usman Mirza,Matheus Froeyen
Department of Pharmaceutical and Pharmacological Sciences,Rega Institute for Medical Research,Medicinal Chemistry,University of Leuven,B-3000,Leuven,Belgium
Keywords:
SARS-CoV-2
COVID-19 outbreak
CoV-Mpro
CoV-Nsp12 polymerase
CoV-Nsp13 helicase
ABSTRACT
Recently emerged SARS-CoV-2 caused a major outbreak of coronavirus disease 2019(COVID-19)and instigated a widespread fear,threatening global health safety.To date,no licensed antiviral drugs or vaccines are available against COVID-19 although several clinical trials are under way to test possible therapies.During this urgent situation,computational drug discovery methods provide an alternative to tiresome high-throughput screening,particularly in the hit-to-lead-optimization stage.Identification of small molecules that specifically target viral replication apparatus has indicated the highest potential towards antiviral drug discovery.In this work,we present potential compounds that specifically target SARS-CoV-2 vital proteins,including the main protease,Nsp12 RNA polymerase and Nsp13 helicase.An integrative virtual screening and molecular dynamics simulations approach has facilitated the identification of potential binding modes and favourable molecular interaction profile of corresponding compounds.Moreover,the identification of structurally important binding site residues in conserved motifs located inside the active site highlights relative importance of ligand binding based on residual energy decomposition analysis.Although the current study lacks experimental validation,the structural information obtained from this computational study has paved way for the design of targeted inhibitors to combat COVID-19 outbreak.
1.Introduction
The ongoing 2019—2020 outbreak of coronavirus disease 2019(COVID-19)[1],as of April 20,2020,has claimed 157,847 lives globally and has been declared as a pandemic.SARS-CoV-2 belongs to the coronaviruses family and is the seventh known human coronavirus(HCoV)from the same family after 229E,NL63,OC43,HKU1,MERS-CoV,and SARS-CoV[1].
Currently,there is no licensed drug or vaccine available for SARS-CoV-2.Although several clinical trials are in progress to test possible therapies,the treatment is focused on the alleviation of symptoms which may include dry cough,fever and pneumonia[2].Following the SARS outbreak,existing antivirals,including nine protease inhibitors[3],nel finavir blocked SARS-CoV-2 replication at the lowest concentration(EC50=1.13 μM,CC50=24.32 μM,SI=21.52).Following this,antiviral efficiency of several FDA-approved drugs has been reported,including remdesivir(GS-5734,Gilead)(EC50=0.77 μM)and chloroquine(EC50=1.13 μM)against a clinical isolate of SARS-CoV-2 in vitro showing potential inhibition at low-micromolar concentration[4].The efficacy of remdesivir is evident from a recent recovery of US patient infected with SARS-CoV-2 after intravenous treatment[5],while chloroquine is being evaluated in an open-label trial(ChiCTR2000029609).Moreover,Ivermectin,a broad spectrum anti-parasitic agent,was reported in vitro activity(~5000-fold reduction)in a model of Vero/hSLAM cells infected with a SARSCoV-2 isolate(Australia/VIC01/2020)[6].Others include Nafamostat(EC50=22.50 μM),Nitazoxanide(EC50=2.12 μM)and Favipiravir(EC50=61.88μM)[4].Recently,hydroxychloroquine has shown to be effective in COVID-19 patients[7]and its efficiency has been reportedly reinforced by azithromycin for virus elimination[8],although no well-controlled,randomized clinical evidence supports azithromycin therapy in COVID-19[9].
CoVs are single-stranded positive-sense RNA(+ssRNA)viruses with 5′-cap and 3′-poly-A tail.The ~30 kb SARS-CoV-2 genome contains at least six open reading frames(ORFs).The first ORF(ORF1a/b)is about two-thirds of the whole genome length and encodes 16 non-structural proteins(nsp1-16).ORFs near 3′-end of the genome encode four main structural proteins including spike(S),membrane(M),envelope(E),and nucleocapsid(N)proteins.Among non-structural proteins,most are known to play a vital role in CoV replication.Structural proteins,however,are important for virion assembly as well as for causing CoV infection.Additionally,specific structural and accessory proteins such as HE protein are also encoded by the CoV genome[10].SARS-CoV-2 maintains~80% nucleotide identity to the original SARS epidemic viruses[11].
To address the current outbreak,the development of widespectrum inhibitors against CoV-associated diseases is an attractive strategy.However,this approach requiresthe identification of a conserved target regionwithin entire coronavirus genus[12,13].On the contrary,all structural proteins including S,E,M,HE,and N proteins among different CoVs have considerable variations as reported[14—16],thus adding more complexity towards identification of SARS-CoV-2 inhibitors.Subsequently,the Nsp12 RNA-dependent RNA polymerase(RdRp),Nsp13 helicase,and main protease(Mpro)or chymotrypsin-like protease(3CLpro)[17]constitute highly conserved regions in non-structural proteins among coronaviruses which can be targeted.
Although no structural data were available for SARS-CoV-2 proteins at the beginning of this study,considering high sequence similarity,SARS-CoV-2 proteins were modeled for rational drug design which can later lead to downstream modification for drug leads.The current study focusses on structure elucidation of a critically important nCoV-Nsp12 polymerase,Nsp13 helicase and Mpro together with virtual screening(VS).The study reports potential hits identified through an integrative VS and molecular dynamics simulation approach.Although the current study lacks experimental validation of proposed hits,structural information along with the identified potential hits may serve as the starting point for structure-guided drug discovery.
2.Methods
2.1.Protein modeling
Homology models of key SARS-CoV-2 proteins including Mpro,Nsp12 polymerase,and Nsp13 helicase were designed using SWISSMODEL[18]as there were no resolved crystal structures available at the time of this study.Later for molecular docking studies,a recently reported co-crystalized Mpro structure was utilized(PDB ID:6LU7).Templates with the highest identity were selected,and respective models were generated.The residues inside the binding pocket were predicted using an analytical tool,COACH meta-server and compared after superimposing various closely related X-ray resolved co-crystallized structures with bound inhibitors.The generated homology models were refined using 20 ns molecular dynamics(MD)simulations to remove steric clashes and optimize side-chain geometry.
AMBER18 simulation package was utilized for unrestrained MD simulations using AMBER ff99SB force field[19].The stepwise minimization and equilibrationprotocol were performed(details in Supplementary information),and solvated system for each model with explicit TIP3 water molecules was submitted to a production run of 20 ns at constant 300K temperature and 1 bar pressure.The protein backbone conformation was analysed using CPPTRAJ module and evaluated through Ramachandhran plots.The most favourable conformation was selected for virtual screening.
2.2.Structure-based virtual screening and comparative docking
Small molecules dataset in SMILES format was downloaded from ZINC database[20]and uploaded to Mcule which is a fully automated drug discovery platform[21].Virtual screening pipeline was established using drug-like filters.Diversity selection was implemented to reduce the size of the library and to maximize the coverage of chemical space at the same time.This procedure excluded the closest analogues based on the Tanimoto similarity coefficient and maximized the diversity of the potential active scaffolds identified from the huge dataset.A thorough analysis of binding site residues was performed prior to docking,as depicted from COACH and structure comparisons.Docking grid was set up covering the key binding site residues to reduce the search space for ligand optimization.
An automated in-built Autodock(AD)Vina docking engine was utilized to screen the composed docking library,which uses gradient optimization method in its local optimization process to efficiently rank the best poses[22].Individual compounds of the docking library together with Mcule database were docked iteratively into respective binding pockets of the representative SARSCoV-2 Mpro,Nsp12 polymerase,and Nsp13 helicase structures.The compounds were ranked based on Vina empirical scoring function that approximates the ligand binding affinity in kcal/mol.Since various docking programs only estimate the real binding affinity,testing different docking programs add reliability.The top hits obtained from the first round of docking were then evaluated with Glide “extra precision”docking mode(Glide XP)as implemented in Schrödinger's Maestro modeling package(a trial version was utilized for this study).SARS-CoV-2 proteins were prepared using protein preparation wizard by adding hydrogen atoms and missing side chains,while short minimization of only the hydrogen atoms was carried out using the OPLS 3 force field.Selected ligands ranked by AutoDock Vinawere prepared using LigPrep as described previously[23].The grid box was generated with dimensions covering the predicted active site residues.Top hits were selected after extensive post-docking analysis by comparing the binding poses predicted from AD Vina and Glide.
2.3.Molecular dynamics simulations and energy calculations
To gain a better understanding of the predicted molecular interactions,the best docked complexes were analysed over a period of 20 ns MD simulations using AMBER 18 simulation package.The antechamber module of AMBER18 was used to generate atomic partial charges for the selected hits.In this study,previously reported MD simulation protocol was followed[23,24](details in Supplementary data).To gain rational insights into the different binding modes and residual contribution,the Molecular Mechanics/Generalized Born Solvent Area(MM/GBSA)method was employed and total binding free energies(kcal/mol)were calculated.The MM/GBSA approach is well detailed in binding free energy calculations[25]for antiviral inhibitors[26,27].
3.Results
3.1.Structural insights of SARS-CoV-2 proteins
In silico modeling work was initiated by analysing Wuhan seafood marketpneumoniavirusgenome(NCBIgenomeID MN908947),which was recently deposited byWu et al.[28].For the currentwork,orf1ab polyprotein sequence (GenBank ID:QHD43415.1)was considered(a total of 7096 amino acids).In order to locate the potential Nsp12,Nsp13 and Mpro sequences,a multiple sequence alignment was performed with known SARS-CoV proteins(Uniprot ID:P0C6X7).The corresponding regions in SARS-CoV-2 genome were identified based on the sequence identity and similarity score.Sequence alignment revealed that SARS-CoV-2 Nsp12 polymerase,Nsp13 helicase and Mpro shared high similarity with that of SARS.
Homology modeling was performed using SWISSMODEL and generated models were validated accordingly.SARS-CoV proteins deposited in Protein Data Bank(PDB)indicated 96.35% ,99.83% and 96.08% identities with SARS-CoV Nsp12(6NUR),Nsp13(6JYT)and Mpro(2Z9J).As a result of high sequence similarity(>96% ),the homology models revealed a strikingly conserved overall architecture.The superimposition of RdRp,helicase and Mpro with SARS-CoV proteins demonstrated high structural similarity,with root mean square deviation(RMSD)values of 1.35 Å,0.25 Å and 0.46 Å,respectively.Model validation using PROCHECK indicated 96.51% (Mpro)and 97.5% (RdRp)of all residues in Ramachandran favoured regions with 3.27% and 0.28% being rotamer outliers,while helicase indicated 84.34% with 4.43% outliers.The models were further refined by 20 ns MD simulation and compared with the templates.With the availability of the crystal structure of SARSCoV-2 Mpro(6LU7)complexed with N3 peptide,homology model was also compared and less than 0.5 Å RMSD was found,which indicated the reliability of the generated model with only subtle difference(Fig.S1-S3).Overall,SARS-CoV-2 Nsp12 polymerase and Nsp13 helicase models were reliable enough to perform virtual screening.For Mpro,co-crystalized structure(6LU7)was utilized.All models revealed the conserved features especially in functional regions due to strikingly similar protein conformation.However,some of the structural features have been elucidated together with the identification of inhibitor binding site in SARS-CoV-2 proteins as shown in Fig.1.
3.1.1.SARS-CoV-2 Mpro
SARS-CoV-2 Mpro was predicted to contain 306 amino acids(located in the polyprotein,from 3264 to 3569 a.a).Structural analysis of SARS-CoV-2 Mpro revealed the same location of the active site as reported in SARS-Mpro,which is arranged in the cleft between domain I(8—99 a.a)and domain II(100—183 a.a).Both domains contribute one residue to the catalytic dyad(His41 and Cys145)(Fig.1A),connected by a long loop(184—199 a.a)to the helical domain III(200—306 a.a).Superimposition of several cocrystalized SARS-Mpro structures bound with inhibitors[13],N1(1WOF),I2(2D2D),N3(2AMQ),and N9(2AMD),also revealed the similarpositionofS1,S2,andS4subsites,especiallyintheactivesite close to His41 and Cys145,which is crucial for substrate recognition[13],along with Tyr161 and His163 in the substrate-binding pocket[29].Docking grid was formed around these subsites for virtual screening by taking co-crystalized SARS-CoV-2 Mpro(6LU7),and top hits were identified based on the docking scores.
3.1.2.SARS-CoV-2 Nsp12 RdRp
The SARS-CoV-2 Nsp12 polymerase was predicted to contain 932 amino acids(located in the polyprotein,from 4393 to 5324 a.a).The nsp12 comprised N-terminal(1—397 a.a)and a polymerase domain(a.a.398—919)when compared with SARS-CoV Nsp12(6NUR).The polymerase domain adopted a structure resembling a cupped “right hand”like other polymerases[24].Nsp 12 polymerase is composed of a finger(398—581 and 628—687 a.a),a palm(582—627 and 688—815 a.a),and a thumb subdomain(816—919 a.a)(Fig.1B).Similar to MERS and SARS-Nsp12,the finger and the thumb subdomains of nCoV-RdRp contacted each other,which configured the RdRp active site in the centre for the substrate access through template entry,template-primerexit,and NTPtunnels[30].Alongside,the SARS-CoV-2 Nsp12 also revealed seven conserved motifs(A—G)arranged in the polymerase active site chamber,which are involved in a template and nucleotide binding and catalysis[31].The binding site of SARS-CoV-2 Nsp12 was further analysed through thesuperimposition ofelongation complex of poliovirus bound with CTP-Mn(3LO8)[32]and crystal structure of Japanese encephalitis RdRp in complex with ATP(4HDH)[33].Docking grid was formed around the polymerase active site chamber covering the conserved motifs(A-G),and potential hits were identified.
3.1.3.SARS-CoV-2 Nsp13 helicase
The SARS-CoV-2 Nsp13 was predicted to contain 596 amino acids(located in or flab polyprotein from5325 to5925 a.a).Like that of SARS and MERS-Nsp13,the overall structure of SARS-CoV-2 Nsp13 adopted a triangular pyramid shape comprising five domains.Among these,two “RecA-like”domains,1A(261—441 a.a)and 2A(442—596 a.a),and 1B domain(150—260 a.a)form the triangular base,while N-terminal Zinc binding domain(ZBD)(1—99 a.a)and stalk domain(100—149 a.a),which connects ZBD and 1B domain,are arranged at the apex of the pyramid(Fig.1C).Small molecules able to inhibit the NTPase activity by interferences with ATP binding may serve as an ideal strategy to develop inhibitors.The SARS-CoV-2 Nsp13 revealed the similar conserved NTPase active site residues including Lys288,Ser289,Asp374,Glu375,Gln404 and Arg567 as present in SARS-Nsp13.All these residues were clustered together in the cleft located at the base between domains 1A and 2A,while docking grid was formed by locating bound ADP of crystalized yeast Upf1 and top hits were identified.
3.2.Identification of potential compounds against SARS-CoV-2 proteins
A stepwise structure-based virtual screening pipeline was adopted considering the putative binding sites of SARS-CoV-2 Mpro,Nsp12 polymerase and Nsp13 helicase to identity potential hits as described in previous studies[24,34,35].The identified potential hits against each target were subjected to in silico ADMET(absorption,distribution,metabolism,elimination,toxicity)predictions,which includes drug-likeness and toxicity potential.This filtering removed substantial hits that exhibited poor ADMET due to inhibitory effects on the renal organic cation transporter and CYP450 isozymes.While a large subset of hits was found to contain high-risk chemical groups like epoxides and quinones,the potential hits against each target with the lowest docking energy were analysed for their binding pose inside the binding site.After careful inspection,best hits based on AD Vina that predicted binding energies were re-docked with Glide and best docked complexes were further processed through 20 ns MD simulations.MMGBSA method was employed to analyse electrostatic and vdW energy contribution to total free energy of binding.The MD-simulated complexes indicating protein backbone stability(during the last 5 ns in a 20 ns production run)were considered for further molecular interaction analysis.Based on molecular interactions and binding free energy calculations,the subsequent screening resulted in 5,4 and 3 hits for SARS-CoV-2 Mpro,Nsp12 and Nsp13,respectively(Detailed ADMET profile of these compounds is tabulated in Table S1,estimated from Swiss-ADME server[36]).The top hits with ADVina,Glide XP-score together with contribution of electrostatic(ΔEele),van der Waals(ΔEvdw),and solute-solvent energies(ΔGnpand ΔGp)towards total binding free energy(ΔGtol)were estimated through Amber-MMGBSA module as tabulated in Table 1.All energy contributions were calculated in kcal/mol.
3.3.SARS-CoV-2 Mpro hits
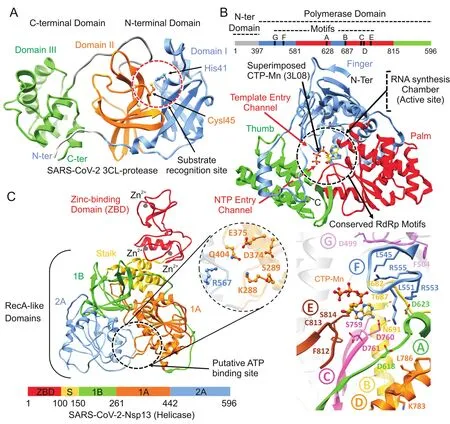
Fig.1.Structural representation of SARS-CoV-2 proteins.(A)SARS-CoV-2 main protease monomer(ribbon representation)composed of:(i)N-terminal domain I(corn flower blue),(ii)domain II(orange),and(iii)C-terminal domain III(green).Substrate recognition site in circle(red)and catalytic dyad residues,His41 and Cys145 are highlighted and labelled.(B)Linear schematic description of domain architecture of SARS-CoV-2 Nsp12 polymerase followed by its structure composed of thumb(green),palm(red),and finger(corn flower blue)subdomains.The active site of Nsp12 polymerase is highlighted and arrangement of structurally conserved RdRp motifs in Nsp12 polymerase model coloured green(A),yellow(B),pink(C),orange(D),brown(E),corn flower blue(F)and magenta(G)for motifs A-G respectively is displayed in bottom right.Superposition of the polio virus elongation complex structure(PDB:3OL8)CTP(orange)inside the predicted binding site also displayed.(C)Overall structure of SARS-CoV-2 Nsp13 helicase composed of ZBD(red),stalk(golden),1B(green),1A(orange)and 2A(corn flower blue)domains.Three zinc atoms in ZBD are shown as dark grey spheres.The binding pocket residues are zoomed in and labelled.Linear schematic diagram of the domain organization of SARS-CoV-2 Nsp13 helicase is displa yed at the bottom.
As a validation docking protocol,the cocrystalized N3 peptide was removed and redocked with the substrate-binding site of SARS-CoV-2 Mpro(6LU7)using the same docking parameters.The generated redocked pose was quite similar to the co-crystalized conformation(Fig.S4).Top-ranked SARS-CoV-2 Mpro hits were filtered based on the interaction with the catalytic dyad(at least one H-bond with either His41 or Cys145 or strong vdW interactions).A total of 13 compounds were analysed after 20 ns MD simulation based on the stability for at least last 5 ns.MMGBSA calculations were performed for the most stable complexes.Among top hits,total binding free energies(ΔGtol)were very promising for cmp12, cmp14, cmp17 and cmp18 which exhibited ΔGtol=-45.22,-44.91,-41.72,and-44.55 kcal/mol.In all complexes,ligand stayed inside the binding pocket and backboneRMSD of Mpro remained stable throughout simulation period(Fig.S5).All these compounds contributed well through H-bonds and exhibited favourable electrostatic energy for cmp3(ΔGele=-21.38 kcal/mol),cmp12(ΔGele=-26.16 kcal/mol),cmp14(ΔGele=-33.56 kcal/mol)and cmp18(ΔGele=-36.65 kcal/mol)except cmp17,which displayed weak electrostatic energy(ΔGele=-5.39 kcal/mol)due to unstable H-bonds over a simulation period(Table 1).Further indepth molecular interaction analysis after 20 ns MD simulations unveiled significant findingswith respectto Mpro-subsites(Fig.2A)and per-residue decomposition is displayed in Fig.2B.Among all compounds,cmp12 and 14 showed some consensus features,where pentacyclic moiety of cmp12 anchored right inside the subsites and formed H-bonds with Ser144 of S1,Cys145 of catalytic dyad,Met165 of S4,and terminal 4-methylaniline established two H-bonds with two neighbouring glutamine residues(Gln189 and Gln192)of S4.Similarly,biphenyl interacted hydrophobically with S2,triazole moiety established one H-bond with Cys145 of catalytic dyad,and pyrazole-pyrimidine moiety formed three H-bonds with Gln189,Thr190,and Gln192 located at S4.The remaining compounds(cmp3,cmp17 and cmp18)demonstrated distinct H-bond patterns,even though the central moieties superimposed well near the catalytic dyad.For instance,the terminal oxadiazol-2-amine moiety of cmp3 mainly interacted through H-bonds with Cys145 of catalytic dyad,Asn142,Ser144,Glu166 andHis172 of S1,and central carbaldehyde also formed H-bond with Glu166 of S1.In contrast,cmp17 only formed H-bonds with residues of S4 through terminal carbazole-1-carboxamide moiety and cmp18 established H-bonds with residues of S2 and S4 through urea and terminal imidazole moieties.Although the compounds mainly interacted electrostatically,nonpolar solvation energies also slightly contributed to ligand binding.The residual decomposition analysis revealed the favourable contribution of catalytic dyad to total binding free energy, where Cys145 exhibited-1.058,-1.19,-2.12,-1.015,and-0.892 kcal/mol and His41 exhibited-0.423,-1.528,-0.656,-1.032,and-0.532 kcal/mol with cmp3,12,14,17 and 18,respectively.Other residues located in close vicinity to catalytic dyad which also contributed significantly included Glu189(ranging from-3.505 to-1.219 kcal/mol),Glu166(-2.711 to-0.845 kcal/mol),and Met165(-3.859 to-0.988 kcal/mol)(Fig.2B).Such favourable residual energy contribution was evident from the H-bonds formed by Glu166,Glu189 and Met165.The 2D chemical structure representation of each compound is displayed in connection with interactions of structural moieties with binding pocket subsites(Fig.2C and D).
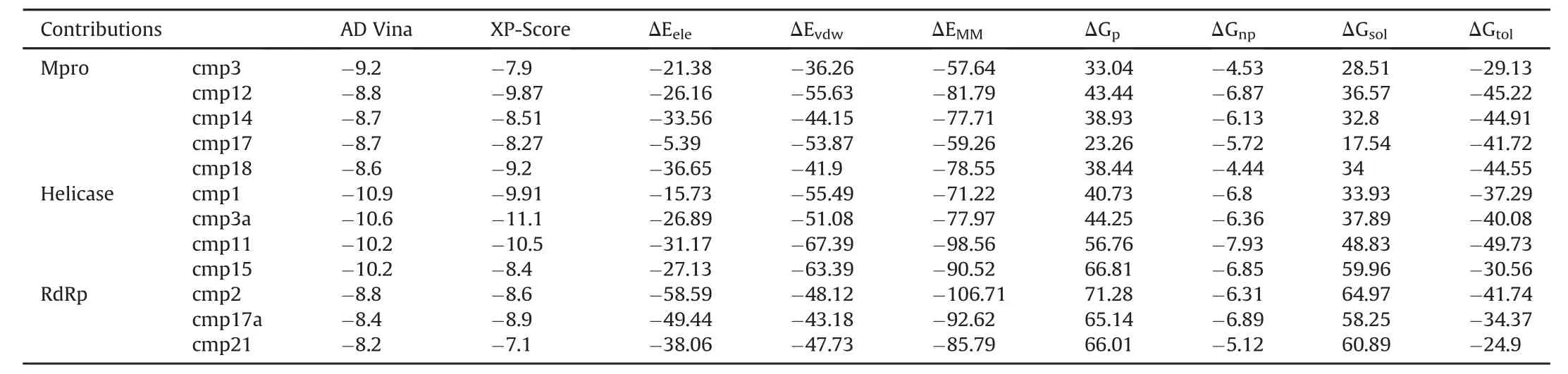
Table 1 Molecular docking predicted binding energies(kcal/mol)and molecular mechanics generalized born surface area(MM-GBSA)binding free energy calculation of identified hits in complex with SARS-CoV-2 Mpro,Nsp12 polymerase and Nsp13 helicase.
3.4.SARS-CoV-2 Nsp12 RdRp
For SARS-CoV-2 Nsp12 RdRp,hits were selected based on establishing H-bond with conserved motifs,especially with signature motif C(SDD),which has been experimentally evaluated in altering polymerase activity supported by various mutational studies[37—39].A total of 17 hits were analysed through 20ns MD simulations,and complexes with stable RMSD were further processed through MMGBSA binding free energy calculations.Among all hits,cmp2,cmp17a and cmp21 showed promisingΔGtol(-41.74,-34.37,and-24.9 kcal/mol)which mainly interacted through H-bonds,thus contributing electrostatically(Table 1).All these complexes remained stable throughout 20 ns MD simulations(Fig.S6).A careful molecular inspection after MD revealed the similar conformation of these compounds,which were found to interact extensively with the conserved motifs(Fig.3).
Cmp2 established three H-bonds with the residues belonging to signature motif C(Ser759),motif D(Lys798)and motif E(Ser814)(Fig.3B),Cmp17a formed five H-bonds with Arg624(motif A),Thr680 and Ser682(motif B),Ser756(motif C)and Lys798(motif D)(Fig.3C),whereas cmp21 interacted mainly with the residues of motif A(Asp623),motif F(Arg553 and Arg555),and motif C(Ser759)through four H-bonds(Fig.3D).Together various residues also contributed through strong hydrophobic interactions.For instance,Tyr455 established strong stalking interaction with terminal difluorobenzene of cmp21.Per-residue binding energy decomposition analysis revealed a key role of important residues located in conserved polymerase motifs(Fig.3F).These include the residues:(i)Arg553 and Arg555 of motif F which exhibited most favourableΔGtolwith cmp2(-2.05 and-2.14 kcal/mol),cmp17a(-1.84 and-2.57 kcal/mol)and cmp21(-2.4 and-1.04 kcal/mol),(ii)conserved aspartate residues of motif A(Asp618 and Asp623)and C(Asp760),(iii)conserved serine residues of motif B(Ser682)and motif C(Ser759),and(iv)Lys798 of motif D.Among these,Ser759 and Asp760 of motif C exhibited significant binding free energy with all compounds(rangingfrom-0.988 to-1.76 kcal/mol and-1.03 to-1.47 kcal/mol),while most of the residues interacted electrostatically.By the end of long-run simulation process,an X-ray resolved SARS-CoV-2 Nsp12 polymerase structure was deposited(6M71)[40].We comparedcrystal structure of SARS-CoV-2 and found only 0.41 Å deviation(Fig.S7).In further support,MD simulations of homology model and 6M71 also showed consensus RMSD over a period of 50ns.Moreover,the conformations of cmp2,cmp17a and cmp21 showed identical binding modes when docked inside the polymerase active site of crystal structure.Additionally,the binding mode and molecular interaction profile of cmp2,cmp17a and cmp21 agreed with the reported comparative binding model of remdesivir(7BV2)and sofosbuvir diphosphate(HCVRdRp,4WTG)[41]with polymerase(Fig.S7)[40].
3.5.SARS-CoV-2 Nsp13 helicase
For SARS-CoV-2 Nsp13 helicase,top hits were selected based on significant interactions with six key residues(Lys288,Ser289,Asp374,Glu375,Gln404 and Arg567)including neighbouring residues involved in NTP hydrolysis[42].A 20 ns MD simulation on top ranked complexes led to the identification of 4 best compounds based on considerable interactions with key residues and demonstrated favourable MMGBSA binding free energy(Table 1).All these complexes remained stable throughout 20 ns MD simulations except cmp3a,which fluctuated in the start and adopted a more favourable conformation inside the binding pocket(Fig.S8).The superimposition of Upf1-ADP showed the overall same binding mode as identified with these 4 compounds(Fig.4).

Fig.2.Post-molecular dynamics(MD)analysis of SARS-CoV-2 Mpro hits.(A)Molecular surface representation of Mpro with MD simulated representative conformation of cmp3(yellow),cmp12(green),cmp14(magenta),cmp17(cyan)and cmp18(dark blue)inside the substrate binding site zoomed with subsites S1(orange),S2(pink)and S4(cyan)and residues are labelled accordingly.Molecular interactions representations of each complex with interacting residues are highlighted in blue sticks and catalytic dyad in green sticks.(B)Per-residue energy decomposition analysis of potential substrate-binding site residues.Terminal moieties of cmp12 and cmp14 revealed similar binding modes while cmp3,cmp17 and cmp18 showing different binding mode are displayed in(C)and(D)followed by chemical structure representations of these compounds in connection with interactions of structural moieties with binding pocket subsites.
Two dimensional(2D)interaction plot of yeast Upf1-ADP revealed a network of H-bonds established by phosphate groups with Lys436,Thr434,Gly433,Arg639 Gly435 which were also conserved in nCoV-helicase(Fig.4A).MMGBSA method estimated favourableΔGtolin cmp1(-37.29 kcal/mol),cmp3a(-40.08 kcal/mol)and cmp11(-49.73 kcal/mol)except cmp15(-30.56 kcal/mol)andcontributedmainlythroughvdW energies,while nonpolar solvation energies also slightly contributed(Table 1).Indepth molecular analysis of cmp11 showed most significant MMGBSAtotal energy,which indicated 3 H-bonds wereestablished between Ser310,Glu375 and Lys288 and terminal region of cmp11,and interacted mainly with the side chains of Glu374,Met378,Ala312,Arg178,Ser535.Ser539 and Asp534(Fig.4B).These residues were also found conserved in Upf1-ADP complex except Met378 and Arg178.Per-residue energy decomposition analysis indicated major contributions from Arg178 of domain 1B,Ala312 and Ala314 of domain 1A,which indicated<-1.5 kcal/mol binding energy with all four compounds.Together,Ser288 and Ser310 also contributed in total free energy of binding(Fig.4C).
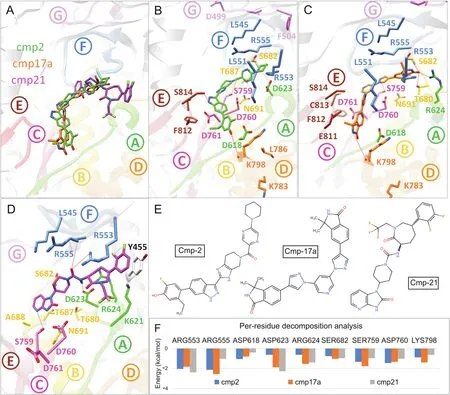
Fig.3.Post-molecular dynamics(MD)analysis of SARS-CoV-2 Nsp12 RdRp complexes.(A)MD simulated conformations of cmp2(green)(B),cmp17a(orange)(C),cmp21(magenta)(D)inside the predicted binding pocket of Nsp12 polymerase.The arrangement of motifs and colors are the same as in Fig.1B.Molecular interactions of individual compounds are displayed in B and C and D,and residues are labelled accordingly.(E)Representations of chemical structures of compounds,and(F)Per-residue energy decomposition analysis of potential binding pocket residues.
4.Discussion
The computational methods offer an immediate and scientifically sound basis to design a highly specific inhibitor against important viral proteins and give aid in antiviral drug discovery[43,44].A recent structural modeling effort predicted few repurposed drugs against SARS-CoV-2 vital proteins including antibacterial,anti-fungal,anti-HIV drugs,and flavonoids from different sources[45].In the current study,a brief structural elucidation was performed together,which revealed strikingly similar features in SARS-CoV-2,when compared with crystal structures of SARS-CoV-RdRp(6NUR)and helicase(6JYT).These viral proteins playa crucial role in the viral life cycle and considered among the most popular strategies for antiviral therapeutics[44,46].Structural analysis has revealed that the substrate-binding pockets of SARS-CoV-Mpro,active site of RdRp and NTPase binding pocket of helicase are highly conserved[47],which can lead to the conceptof “wide-spectrum inhibitors” fortargeting CoVs.Furthermore,extensive SBVS procedure led to the identification of potential hits against each target,which were assessed through MD simulations.MM-GBSA and per residue energy decomposition divulged the relative importance of amino acid involved in binding supported with the contribution of different components of binding free energy.
Overall,SARS-CoV-2-Mpro hits were found to interact with catalytic dyad(His41 or Cys145)together with residues located at S4,S2 and S1 subsites of substrate binding pocket and mutational studies of His41 or Cys145 resulted in complete loss of enzymatic activity,which confirmed the role of catalytic dyad[48,49].In the substrate-binding pocket,catalytic dyad residues,highly conserved residues of S1,Gln189 and Met165 of S4 displayed relative importance in ligand binding,suggesting a possibility for inhibitor design targeting this region in the Mpro.In case of SARS-CoV-2 Nsp12,the in-depth interaction analysis inside the conserved motifs(Fig.3)revealed functionally important aspartate residues of motif A(Asp623)and motif C(Asp760),together with conserved arginine residues of motif F(Arg553 and Arg555),and Ser759(motif C)which were found to highly interact with the compounds.Among these,motif A and C are most strikingly conserved aspartate residues that bind divalent metal ions necessary for catalytic activity[30,31].Moreover,the importance of aspartate residues in RdRps supported by various other mutational studies[37—39]suggested a conserved binding feature of cmp2,cmp17a and cmp21 along with side chains of Arg553,Arg555 and Ser759 which lie deep in the binding pocket of nCoV-RdRp,thus enhancing it as an anchor for inhibitory molecules.

Fig.4.Post-molecular dynamics(MD)analysis of SARS-CoV-2 Nsp13 helicase complexes.(A)Yeast Upf1-ADP complex(transparent white)superimposed on Nsp13 helicase to locate the ATP putative binding pocket and 2D-interaction plot is displayed.MD simulated cmp1(brown),cmp3a(magenta),cmp11(green)and cmp15(sandy brown)are displayed inside the pocket.The arrangement of domains is coloured the same as in Fig.1C.(B)MD simulated conformations of cmp11(green)inside the predicted binding pocket of Nsp13 helicase interacted with key residues of domain 1A,2A and 1B.(C)Per-residue energy decomposition analysis of potential binding pocket residues,and representations of chemical structures of compounds are displayed in(D).
In SARS-CoV-2-helicase,the structural information of NTPase active site obtained after superimposing SARS-helicase and yeast-Upf1-ADP divulged the location of conserved residues.The identified hits were positioned similarly with reference to the interaction network reported for co-crystalized ADP(Fig.4).In the active site,the side chains of Ser310,Lys288 and Glu375 actively participated in H-bond,while Arg178 and conserved alanine residues(Ala312 and Ala314)contributed substantially to total binding free energy.Moreover,the importance of these residues has already been highlighted in mutational studies[42].
Additionally,the identified compounds exhibited promising ADMET properties and none of the compounds were found to contain any high-risk chemical group.The drug candidates with poor ADMET might lead to toxic metabolites[50]or be unable to cross membranes[51],and the predicted solubilities were estimated within an acceptable range[52].Most of the compounds were predicted as non-inhibitors for cytochrome P450 isozymes which play a vital role in drug metabolism and inhibition of these enzymes leads to toxicity and poor excretion from the liver[53].Moreover,all compounds were found novel as they were not reported elsewhere against antiviral activities.Among various hits identified,cmp2 against SARS-CoV-2 RdRp was recently reported as a Pan-Janus Kinase Inhibitor Clinical Candidate(PF-06263276)for its potential role in the treatment of inflammatory diseases of lungs[54].Hence,the cmp2 could be ideal for reducing the inflammatory activity during pathogenic COVID-19 epidemic,as lung inflammation has been observed during the SARS and MERS outbreaks.
Conclusively,this comprehensive study presents an integrated computational approach to the identification of novel inhibitors into the area of anti-nCoV treatment as a starting point,which warrants in vitro testing to evaluate compound efficacy.The indepth structural elucidation of interacting residues together with the dynamic conformations adopted over a period of 20 ns MD simulations of identified compounds offers the way to designwidespectrum or selective inhibitors against HCoV.
Compliance with ethics requirement
This article does not contain any study with human or animal subjects.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgments
We would like to thank the computational facilities provided by KU Leuven.This study was supported by IRO scholarship Ph.D.Grant.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2020.04.008.
 Journal of Pharmaceutical Analysis2020年4期
Journal of Pharmaceutical Analysis2020年4期
- Journal of Pharmaceutical Analysis的其它文章
- Plant-derived secondary metabolites as the main source of efflux pump inhibitors and methods for identification
- Polysaccharide-based chromatographic adsorbents for virus purification and viral clearance
- Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants
- Rapid detection of high-risk HPV16 and HPV18 based on microchip electrophoresis
- Solid and liquid state characterization of tetrahydrocurcumin using XRPD,FT-IR,DSC,TGA,LC-MS,GC-MS,and NMR and its biological activities
- Identification of impurities in nafamostat mesylate using HPLC-ITTOF/MS:A series of double-charged ions