
Genetic targeting of astrocytes to combat neurodegenerative disease
2020-09-18 07:04RachelryAllenChenGregoryKirschen
中国神经再生研究(英文版) 2020年2期
Rachel Kéry , Allen P. F. Chen , Gregory W. Kirschen
1 Medical Scientist Training Program (MSTP), Stony Brook Medicine, Stony Brook, NY, USA 2 Department of Neurobiology & Behavior, Stony Brook University, Stony Brook, NY, USA
Abstract Astrocytes, glial cells that interact extensively with neurons and other support cells throughout the central nervous system, have recently come under the spotlight for their potential contribution to, or potential regenerative role in a host of neurodegenerative disorders. It is becoming increasingly clear that astrocytes,in concert with microglial cells, activate intrinsic immunological pathways in the setting of neurodegenerative injury, although the direct and indirect consequences of such activation are still largely unknown. We review the current literature on the astrocyte’s role in several neurodegenerative diseases, as well as highlighting recent advances in genetic manipulation of astrocytes that may prove critical to modulating their response to neurological injury, potentially combatting neurodegenerative damage.
Key Words: Alzheimer's disease; amyotrophic lateral sclerosis; glia; immune system; inflammation; Parkinson's disease; reactive astrocyte; regeneration
Astrocytic Response to Neurodegeneration
Astrocyte signaling and changes in cellular behavior have been associated with a wide variety of neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) (Pehar et al., 2017), Parkinson’s disease (PD) (Miyazaki and Asanuma, 2017), Huntington’s disease (Hsiao et al., 2013),and Alzheimer’s disease (AD) (Assefa et al., 2018). Whether these changes represent a protective/restorative response or a compounding, injurious role is still a matter of controversy.Astrocytes secrete a variety of soluble small molecules and proteins which likely contribute both to normal neural functioning and to the central nervous system (CNS) response to injury (Jha et al., 2018). However, the precise functions of many of these astrocytic-secreted factors remain unclear (Jha et al., 2018).
Astrocytes respond to neural injury or disease through complex and heterogeneous changes in gene expression and glial cell function known as reactive gliosis (Anderson et al.,2014). Reactive gliosis is necessary for proper recovery from brain injury (Anderson et al., 2014; Pekny et al., 2014; Choi et al., 2018), but can also have damaging effects (Anderson et al., 2014; Pekny et al., 2014). A wide variety of signaling pathways and mechanisms can induce astrocytes to initiate an immune response, including local hypoxia (Chavez et al.,2006; Badawi et al., 2012; Huang et al., 2014; Ramamoorthy et al., 2019), mast cell infiltration (Kempuraj et al., 2017;Kempuraj et al., 2019), microglial signaling (Kirkley et al.,2017), myelin debris (Ponath et al., 2017), and inflammation/growth factors released by damaged neurons (Cassina et al., 2005; Gontier et al., 2015). Damaged neurons themselves release microRNAs and other factors also capable of driving astrocytes toward an inflammatory response (Hoye et al., 2018).
Protein aggregation, a common feature in various neurodegenerative disorders, can mediate astrocyte activation(Kovacs et al., 2017). While astrocytes are generally more resistant to the oxidative stress of misfolded protein aggregation as compared to neurons (Zhao et al., 2017), the accumulation of such proteins can interfere with healthy astrocyte functions (Croisier and Graeber, 2006; Fellner et al., 2011; Lim et al., 2018), and lead to astrocytic-mediated neuroinflammation (Sankar et al., 2018). Reactive astrocytes cluster together around amyloid plaques and tau tangles in post-mortem tissue samples taken from AD patients (Serrano-Pozo et al., 2011). In healthy murine astrocytes, the over-accumulation of amyloid-β (Aβ) triggers an astrocytic immune response (Sankar et al., 2018). Fibrinogen plaques,a common feature in AD, multiple sclerosis, stroke, and traumatic brain injury, can also mediate astrocyte activation(Clark et al., 2018). Similarly, the aggregation of α-synuclein,a key step in the pathogenesis of PD, can be directly passed from neurons to astrocytes, triggering an astrocyte-mediated inflammatory response (Lee et al., 2010a, b). Mice that over-express a PD-associated mutant form of α-synuclein in an astrocyte-specific manner have been shown to display Parkinsonian motor symptoms (Cu et al., 2010). Analogously, expression of mutant huntingtin protein in murine astrocytes leads to an age-dependent recapitulation of certain aspects of Huntington’s disease symptomatology (Bradford et al., 2009). These data taken together suggest that astrocyte-mediated neuroinflammation, triggered by protein aggregation may be an important potential therapeutic target for treating neurodegenerative disorders. While astrocytes are not the resident immune cells of the CNS, a role canonically attributed to microglia, astrocytes do utilize immune signals in response to perceived stressors and protein aggregates in a complex fashion.
This review was compiled via PubMed and Google Scholar searches, without date restrictions. Selection criteria included articles written in or translated into English, pertaining to or including the terms astrocyte, glia, neurons and/or inflammation, and/or neuroinflammation, and/or neurodegeneration, and/or genetic manipulation.
Consequences of Astrocyte Activation
Astrocytes respond to inflammatory challenges using many of the same signaling cascades utilized by bona fide myeloidand lymphoid-lineage cells, including microglia. In the course of responding to the insult, astrocytes may lose functions important for supporting healthy neurons and synapses, while gaining abnormal functions that can have negative consequences for neural survival and activity (Sofroniew,2009). However, not all astrocytic transcriptional changes in response to neurodegeneration are necessarily maladaptive,for example clearing debris, and containing protein aggregates within a particular area (Pekny et al., 2007; Sofroniew,2009; Anderson et al., 2014; Hoshi et al., 2018).
Astrocytic responses to inflammation
Astrocytes respond to inflammatory challenges with a wide variety of gene expression changes, dependent on animal age(Kluge et al., 2018), systemic immune status (Rakic et al.,2018), and the specific immune trigger (Perriot et al., 2018).One of the most common pathways activated by immune insult is the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) immune signaling cascade (Kempuraj et al., 2017, 2019). NFκB-mediated astrocyte dysfunction can initiate neurodegeneration through different mechanisms(Lattke et al., 2017; Kim et al., 2018), including astrocytic release of apoptotic factors (Kia et al., 2018), and cytokines/chemokines. These signals orchestrate bidirectional interactions between astrocytes and microglia in the context of neuroinflammation.
Immune-reactive astrocytes produce a number of cytokines and chemokines which can activate the local microglia.For instance, under conditions such as expression of mutated ALS-associated protein, reactive astrocytes can release tumor necrosis factor-α (TNF-α) (Kia et al., 2018), which can activate microglia, driving them toward a pro-inflammatory(M1) phenotype (Boche et al., 2013). Astrocytes can also synthesize lactosylceramide and interleukin-6, other pro-inflammatory drivers of microglia (Mayo et al., 2014; Savarin et al., 2015). Astrocytic production of interleukin-12 can induce interleukin-7 expression in both human and murine microglia (Jana et al., 2014), which, in turn, can amplify lymphocyte proliferation (Lin et al., 2017). T-cell lymphocytes can in turn secrete interferon-γ (Savarin et al., 2015), which enhances inflammatory microglial recruitment and proliferation (Subramaniam and Federoff, 2017). Reactive astrocytes tend to increase their nitric oxide (NO) production (Cassina et al., 2002; Xie and Yang, 2015), which is needed to prime microglial cytokine/chemokine release (Xia and Zhai, 2010),and trigger vasodilation and subsequent immune cell infiltration (Xie and Yang, 2015). Thus, astrocytes triggered by an inflammatory insult, participate actively in the recruitment of resident microglia to sites of injury.
The relationship between reactive astrocytes and microglia is far from one-sided, however, with astrocyte-activated microglia feeding back onto the astrocytes to modulate the extent of the inflammatory response. For instance, activated microglia utilize the C-C chemokine receptor-5 signaling pathway to regulate astrocytic neurotransmitter production,which may be relevant in the context of AD and possibly sporadic PD (Huerta et al., 2004; Xia and Zhai, 2010; Choi et al., 2013; Sahin-Calapoglu et al., 2016). Additionally, anti-inflammatory (M2) microglia produce interleukin-10, which upregulates astrocytic production of anti-inflammatory factors, including transforming growth factor (TGF)-β, which promotes the M2 microglial phenotype (Norden et al., 2014),as well as assisting in neuroprotection. More specifically, one of the principal roles of reactive astrocytes in the context of inflammation is to protect the integrity of synapses. For instance, astrocyte-derived TGF-β1 has been shown to protect synapses from the potentially damaging effects of Aβ aggregation in cultured murine hippocampal neurons (Diniz et al., 2017). Reactive astrocytes may also increase their release of neurotrophic factors, which have been shown to protect cortical synapses against the harmful effects of Aβ aggregation (Guo and Mattson, 2000). Astrocytic TGF-β signaling can also limit the extent of inflammation in response to cerebral infections, such as that caused by the parasite Toxoplasma gondii (Cekanaviciute et al., 2014), and following stroke(Cekanaviciute et al., 2014). The efficiency of astrocyte-microglia signaling appears to dwindle with age, however, perhaps helping to explain the onset of many neurodegenerative diseases later in life. The Figure 1 demonstrates the bidirectional relationship between astrocytes and microglia in the setting of an inflammatory/neurodegenerative insult.
As evidence of this changing cell-cell interaction over time, in aged murine brains, astrocytes have decreased responsiveness to inter leukin-10, leading to prolonged microglial activation following a peripheral immune insult(Subramaniam and Federoff, 2017). TGF-β expression is also elevated in post-mortem human brain tissue samples taken from patients with AD relative to healthy controls, again implicating it as a marker of immunological stress in the brain and potentially of disease severity as well (Zhang et a.,2016). On the other hand, microglia can also increase astrocyte reactivity. In response to increases in NO production by reactive astrocytes, microglia produce interleukin-1β (Sudo et al., 2015), which can further activate astrocytes, increase astrocytic NO production (Hu et al., 1995), and promote astrocytic migration to the site of injury (Yang et al., 2015).Interleukin-1β also upregulates glial fibrillary acid protein(GFAP) expression and subsequent glial scarring (Sticozzi et al., 2013). There is evidence to suggest that this cross-talk could be maladaptive. For instance, blocking such microglial-induced astrocyte conversion has been shown to be neuroprotective in transgenic mice with the familial PD-associated A53T α-synuclein mutation (Yun et al., 2018).
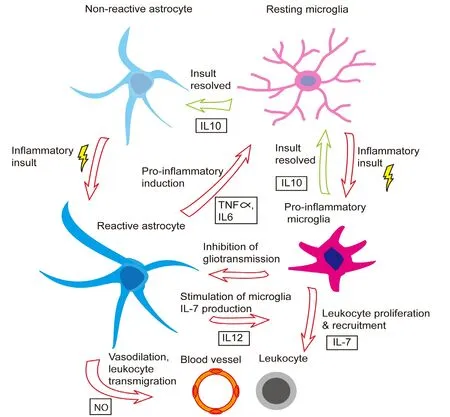
Figure 1 Bidirectional interaction between astrocytes and microglia in regulating neuroinflammation.
Apart from their control by microglia, astrocytes exhibit several other modes of regulation. Peripheral immune cells,such as regulatory T lymphocytes, also regulate astrogliosis(the formation of a scar comprised of reactive astrocytes and other glia and inflammatory cells that borders a focus of inflammation) and the neuro-immune response (Sofroniew,2009; Ito et al., 2019). Moreover, reactive astrocytes are capable of auto-regulation. For instance, in response to oxidative stress, astrocytes upregulate their expression of toll-like receptor-3 which upregulates neuroprotective anti-inflammatory cytokines like interleukin-10, and reduces production of pro-inflammatory cytokines like interleukin-12 (Bsibsi et al.,2006). Reactive astrocytes can also produce neuroprotective lipoxins in response to injury (Livne-Bar et al., 2017).
What factors determine whether astrocytes will assume a pro- or anti-inflammatory phenotype? Astrocyte-derived interferon-1β production appears to be important for this fate determination. Upregulating the expression of interferon regulatory factor 3, a transcription factor required for astrocyte interferon-1β production inhibits astrocyte inflammatory gene expression by suppressing pro-inflammatory microRNAs (Tarassishin et al., 2011). Astrocyte interferon-1 production is known to regulate immune responses of the endothelial cells of the blood-brain barrier, and is associated with anti-inflammatory effects in viral infections of the CNS, and in multiple sclerosis in humans (Rothhammer et al., 2016; Daniels et al., 2017). However, in post-mortem AD brains, upregulation of astrocytic interferon-1 signaling has been associated with promotion of a pro-inflammatory signaling cascade (Taylor et al., 2018). Reactive astrocytes also regulate the peripheral cytokine/chemokine responses by releasing peroxisome proliferator-activated receptor α (Dickens et al., 2017).
The chronicity of CNS inflammation appears to play an important role in determining astrocyte activation status.Chronic activation, for example in the setting of neurodegeneration, increases astrocytic responses to immune insult(Hennessy et al., 2015). In addition, some neurodegenerative diseases are associated with mutations in key astrocyte genes which interrupt astrocytic autoregulation of the neuro-immune response. For instance, fyn tyrosine kinase mutations associated with AD can facilitate persistent inflammatory responses (Lee et al., 2017), and increased microglial inflammatory activity (Panicker et al., 2015). Mutations in the gene for TGF-β have been associated with AD risk (Caraci et al.,2012, 2018), suggesting that defects in astrocytic regulation of microglia may enhance neurodegeneration. Conversely,familial-PD loss-of-function mutations in PTEN-induced putative kinase (PINK1) (Valente et al., 2004) appear to have anti-inflammatory effects (Sun et al., 2018). However, PINK1 loss also induces mitochondrial damage, increasing metabolic stress (Valente et al., 2004). Increased PINK1 expression has been reported in AD and multiple sclerosis lesions(Wilhelmus et al., 2011), where it is thought to stimulate interleukin-1β signaling (Sun et al., 2018). On the other hand,frameshift mutations of the ubiquitin gene, seen in aging and AD-brains can actually suppress inter leukin-1β/TNF-α-mediated astrocyte activation (Choi et al., 2013). Aside from their role in immune modulation, astrocytes assume fundamental functions in CNS metabolic homeostasis.
Metabolic support
A major astrocyte function is the provision of metabolic support for neurons, which is critical in limiting the extent of neuronal oxidative stress (De Miranda et al., 2018). Such support is controlled principally by neurotransmitters (especially glutamate), but also by other small molecule regulators of synaptic signaling including cytokines. Breakdown of metabolic regulation, as may occur in the context of neurodegeneration, highlights the key role of astrocytes in this critical housekeeping function.
Under physiological conditions, astrocytes sense synaptic glutamate levels and use this as a surrogate for neuronal activity, in turn recruiting nutrients and oxygen to metabolically active areas while shunting blood flow away from less metabolically active regions (Bélanger et al., 2011). On the other hand, when the CNS experiences an inflammatory stressor, astrocytes undergo a series of cellular changes to divert their resources to dealing with the stressor. Astrocytic activation causes these cells to neglect their neuro-supportive roles as they instead gear their activity toward inflammatory cell recruitment and glial scar formation (Allaman et al., 2010; Fuller et al., 2010; Steele and Robinson, 2012). For example, in the context of a superoxide dismutase 1 (SOD1)mutant mouse model of ALS, NFκB signaling regulates both the expression of oncogene astrocyte elevated gene-1 and excitatory amino acid transporter-2, decreasing astrocyte clearance of synaptic glutamate in times of oxidative stress(Vartak-Sharma et al., 2014; Yin et al., 2018). This forces neurons to slow down their metabolic activity or else suffer the consequences of oxidative stress.
Fortunately, compensatory mechanisms exist to ensure that neurons decrease their metabolic demand for the purpose of neuronal preservation in the context of oxidative stress. Anti-inflammatory cytokines such as interleukin-10 alter intracellular Ca2+responses to ischemia, preserving neuron viability in the setting of heightened metabolic demand (Tukhovskaya et al., 2014). Following an inflammatory insult, astrocytes also upregulate expression of aquaporin-4(AQP4), and down-regulate expression of glutamate transporter-1 to reduce neuro-excitability (Ikeshima-Kataoka,2016; Lan et al., 2016), thereby reducing cellular metabolic activity. Additionally, interleukin-1β, which is known to activate astrocytes (Xia and Zhai, 2010), also upregulates astrocyte glutathione synthesis, protecting astrocytes (He et al.,2015) and neurons (Chowdhury et al., 2018) from oxidative stress. Mild astrocytic oxidative stress appears to be beneficial, helping to decrease astrocyte inflammatory activation and resultant blood-brain barrier hyper-permeability (Wang et al., 2018).
However, many of the inflammatory factors released as the result of astrogliosis largely have negative consequences for astrocyte metabolic support. For example, interferon-α,released as a consequence of activation, can negatively regulate astrocyte growth and glucose metabolism (Wang et al.,2012). Pro-inflammatory factors released by lymphocytes(interferon-γ) and microglia (TNF-α) can stimulate production of Aβ, potentially increasing plaque load in AD (Yamamoto et al., 2007; Zhao et al., 2011). Astrocytic activation additionally leads to increased production of new astrocytes(Hernández-Guillamon et al., 2009), but also increases astrocyte apoptosis (Saha and Biswas, 2015), potentially exacerbating oxidative/metabolic stress. Abnormal astrocyte metabolism can, in turn, modulate neuroinflammatory responses. For instance, PARK7, a regulator of astrocyte metabolism found to be mutated in some cases of familial PD(Bandopadhyay et al., 2004) is important for astrocyte mitochondrial function (Larsen et al., 2011). Loss of functional PARK7 increases intracellular protein accumulation and subsequent oxidative stress (Kumaran et al., 2007). PARK7 is also important for neural repair, as it helps to actively promote astrogliosis by stabilizing Sox9, a positive regulator of astrogliosis (Choi et al., 2018). An analogous process likely occurs in AD pathology, wherein mutations in the astrocyte protein apolipoprotein E4 can impair Aβ clearance (Liu et al., 2013; Prasad and Rao, 2018), and may increase oxidative stress by impairing the glia-neuron lactate shuttle, leading to an inflammatory response (Liu et al., 2015). In summary,astrocytes not only ensure that nutrient and oxygen supply and demand are balanced at baseline, but are also essential in mediating decreased neuronal energy expenditure in times of oxidative stress for cellular preservation. Aside from their critical role in energy budgeting under neurodegeneration-inducing stress, astrocytes have the added task of ensuring efficient synaptic transmission and are key regulators of neuroplasticity, which are also likely relevant in neurodegeneration.
Synaptic transmission and plasticity
Astrocytes contribute a great deal to the regulation of synaptic transmission and plasticity. In this regard, astrocyte contributions are multifaceted, regulating cholinergic and endocannabinoid synaptic signaling (Navarrete et al., 2012,2014; Gómez-Gonzalo et al., 2015), setting thresholds for long-term potentiation and long-term depression (Bonansco et al., 2011), and regulating neurotransmitter release probability (Perea and Araque, 2007). In fact, the loss of appropriate synaptic and plasticity regulation due to astrocyte activation may help to explain some of the behavioral manifestations of neurodegenerative disorders, for example, the sleep disturbances seen in patients with AD (Vanderheyden et al., 2018).
Again, astrocyte regulation of synaptic transmission appears to be intimately connected to the immunological and metabolic statuses of the CNS. Astrocytes are known to release neurotransmitters such as glutamate at the synapse in a process known as gliotransmission (Halassa et al., 2007).Glutamatergic gliotransmission can be adversely affected by the increased oxidative stress frequently seen in neurodegenerative diseases (Liu et al., 2006). Additionally, inflammatory factors like histamine can trigger increased astrocytic glutamate release (Kárpáti et al., 2018), which could have a potentially negative impact on neural survival. TNF-α, an important astrocyte inflammatory mediator is also important for regulating glutamatergic gliotransmission (Santello et al., 2011). Therefore, it is possible that over-production of TNF-α by astrocytes responding to immune insult could alter gliotransmission.
Aside from neurotransmitter regulation, ion flux is also a key role of astrocytes, the dysregulation of which can exacerbate inflammatory or cytotoxic insults. Astrocytic potassium homeostasis is key for regulation of synaptic plasticity (Djukic et al., 2007), with disruptions potentially contributing to oxidative stress and neurodegeneration. For instance, decreased expression, or loss of expression of the potassium channel Kir4.1 interferes with astrocytic glutamate uptake (Djukic et al., 2007; Kucheryavykh et al., 2007).Glial Kir4.1 expression is progressively lost in mice lacking SOD1, a well-established mouse model for a subset of genetically-linked ALS (Kaiser et al., 2006; Bataveljić et al., 2012),directly implicating astrocytic synaptic dysfunction in this pathological state (Benkler et al., 2013). Activated astrocytes in the spinal cord of SOD1 mutant mice also produce excessive endothelin-1 which can induce excitotoxicity in motor neurons due to over-activation of AMPA receptors (Ranno et al., 2014).
Growth factor signaling
Astrocytes produce a wide array of growth factors, including nerve growth factor, brain-derived growth factor (BDNF),and fibroblast growth factor which are all important for neuron health and survival (Miyazaki and Asanuma, 2016).Some of these astrocyte-produced growth factors are important for maintaining healthy neuronal metabolism by activating anti-oxidative enzymes to reduce over-production of reactive oxygen species (Cabezas et al., 2019). Others help support healthy synapse function: astrocytic fibroblast growth factor supports axon and dendrite outgrowth (Le and Esquenazi, 2002) and BDNF is a well-known regulator of synaptic plasticity (Bramham and Messaoudi, 2005). Still others help to support neuron survival and neurogenesis(Palmer and Ousman, 2018).
Astrocyte production of these growth factors may decrease with age, which may partially account for age-associated increases in incidence of neurodegenerative disorders (Palmer and Ousman, 2018). Upon activation, astrocytes may fail to produce the necessary growth factors to maintain the local neuron population for which they are responsible. For example, mice with the ALS-associated G93A SOD1 mutation harbor reactive astrocytes that fail to produce sufficient nerve growth factor to allow for motor neuron survival(Pehar et al., 2004). Further, changes in serum BDNF levels have been associated with cognitive impairment in patients with AD, frontotemporal dementia, Lewy body dementia,and vascular dementia, potentially implicating these growth factors in these neurodegenerative diseases, although their source is still to be determined (Budni et al., 2015). There is evidence that loss of BDNF in the hippocampus may be associated with various cognitive and emotional symptoms of AD, presumably related to a disruption in healthy experience-driven synaptic plasticity (Budni et al., 2015). In summary, astrocyte-derived growth factors may guard against neurodegeneration by promoting neuronal survival/viability,although more direct evidence will be needed to further establish a causative role of astrocyte-specific growth factor deficiency in neurodegeneration, as well as the therapeutic potential of growth factor therapy in neuroprotection for this purpose.
Conclusions
Astrocytic respo nse to neurodegeneration is diverse, with both positive and negative effects (Sofroniew, 2009; Pekny et al., 2014; Ferrer, 2017). In part this may be due to variability within the overall astrocyte population. There is abundant evidence from transcriptomic and morphological data to suggest that multiple astrocytic subtypes likely exist (Jha et al., 2018; Kirschen et al., 2018; Neal and Richardson, 2018).For instance, consistent with astrocytes’ known interaction with the immune system given inflammatory signals, microarray data have revealed that astrocytes assume a pro-inflammatory transcriptional profile when exposed to cytokine or other soluble molecular signals (Michelucci et al., 2016).Still, more investigation will be required before definitive subcategories of astrocytes can be created. Additionally,astrocyte response to activation can vary with age (Jiang and Cadenas, 2014; Bellaver et al., 2017; Kluge et al., 2018),systemic immune status (Rakic et al., 2018), and depending on the specific trigger for activation (Perriot et al., 2018). A full-understanding of astrocyte transcriptomic and proteomic profiles will be needed in order to determine which targets are most likely to contribute to, versus aid in recovery from neurodegeneration. Despite the many unknowns, astrocytes do appear to be a potentially viable target for precision medicine and may have immense and hitherto largely untapped therapeutic potential (Allen et al., 2017; Barreto et al., 2017).
Potential Therapeutic Targets
Given the wide variability of astrocytes’ responses to inflammation, and consequences of astrogliosis, it becomes challenging to determine which, if any of the mediators of astrogliosis might produce beneficial effects as a therapeutic target for neurodegenerative disease. For example, astrocytic release of TGF-β has been shown to have anti-inflammatory effects: dampening microglial activation (Norden et al.,2014) and potentially limiting the negative synaptic effects of protein accumulation (Diniz et al., 2017). However, constitutive over-expression of TGF-β in mice promotes ADlike microvascular degeneration, suggesting that long-term over-production of TGF-β may ultimately have detrimental effects such as neuronal apoptosis and excessive extracellular matrix protein deposition (Wyss-Coray et al., 2000; Zhang et al., 2016).
It may be more beneficial, therefore, to attempt to target the initiation of the astrogliosis response. There are several potential therapeutic targets that might help to ameliorate protein-accumulation-mediated astrocyte activation. The presence of heme and hemoglobin modulates the astrocytic inflammatory response to the over-accumulation of Aβ in otherwise healthy murine astrocytes, specifically by upregulating phosphoinositide 3-kinase/Akt pathway, ultimately increasing activity of phagocytes via cytokine-mediated recruitment (Sankar et al., 2018). By contrast, in the substantia nigra, activation of astrocytic nicotinic acetylcholine receptors can dampen astrocytic reactivity to immune insults by inhibiting release of cytokines such as TNF- α and interleukins 1β, 2 and 6 (Jurado-Coronel et al., 2016).
Toll-like receptor 2, a protein frequently over-expressed in post-mortem tissue taken from patients with PD and Lewy body dementia (Doorn et al., 2014; Dzamko et al.,2017), may mediate the transmission of α-synuclein between neurons and glia (Kim et al., 2018), and appears to trigger microglial activation through the NFκB pathway (Kim et al.,2016). Astrocytic NFκB signaling may also be modulated by estrogen receptor signaling (Mitra et al., 2016), and by the oncogene astrocyte elevated gene-1 (Vartak-Sharma et al.,2014; Yin et al., 2018). Disrupting astrocytic NFκB signaling greatly reduces astrocytic release of cytokines/chemokines,and subsequent activation (Davis et al., 2015). Thus, a number of potentially druggable pathways exist to attempt to modulate astrocyte immunological function.
It is also desirable to be able to better regulate certain consequences of astrogliosis, as such scarring can greatly limit the degree of potential regeneration after traumatic injury(Pekny et al., 2007). While astrocytes are by no means the only cell type involved in glial scarring, they are integral to glial scar formation. Astrocytic proteins vimentin (VIM) and GFAP are known to modulate transcriptional responses in astrocytes following immune insult (Kamphuis et al., 2015),and are important for glial scar creation (Wilhelmsson et al.,2004). Deleting VIM and GFAP in mice prevents astroglial process hypertrophy, greatly improving post-traumatic regeneration (Wilhelmsson et al., 2004). However, in transgenic mice expressing AD-associated mutant proteins amyloid precursor protein and presenilin-1, loss of GFAP and VIM lead to major increases in Aβ plaque load and neuron dysfunction, suggesting that VIM and GFAP-induced astrocyte activation and subsequent glial scarring may help to contain and limit Aβ plaque growth (Kraft et al., 2013). Given that,whether targeting of glial scar formation serves as a fruitful approach to combatting neurodegeneration remains unclear.
Genetic Strategies for Targeting Astrocytes
Genetic technologies are emerging as a method to target the CNS both for basic studies and for clinical intervention.Here, we will discuss how genetic strategies have been used to understand astrocytes and how they may hold promise for targeting astrocytes in neurodegenerative disease. These strategies allow for specific targeting of astrocytes without affecting other CNS cell types, although various challenges,limitations, and optimization detailed below will be required.Given astrocytes’ regulation of neurotransmitter and ion cycling, neuroplasticity, and neuronal growth and survival,it has been posited that astrocytes may be key to improving cognitive function in the setting of neurodegenerative diseases (Dallérac and Rouach, 2016). Indeed, there is evidence for example that pathological cellular damage induced by excitotoxicity in ALS may be partially attributable to astrocyte expression of GLT1 (Fumagalli et al., 2008). Thus, it stands to reason that experimental manipulation of astrocytes may,under certain circumstances, be able to reverse if not slow the molecular underpinnings of neurodegeneration with the hopes of restoring or preventing cognitive decline among other functions.
To this end, researchers have made large advances in this regard by developing viral gene constructs and transgenic mouse lines that are astrocyte-specific (Shigetomi et al.,2010, 2016; Tsai et al., 2012). The basis of these developments stand on efforts made to define what an astrocyte actually is in terms of the cell’s genetic and morphological features (Khakh and Sofroniew, 2015). Crucial transcriptomic studies have demonstrated that astrocytes express particular genes that are not highly expressed in other CNS cells such as neurons or oligodendrocytes (Gong et al., 2003; Lovatt et al., 2007; Cahoy et al., 2008; Allen and Eroglu, 2017). Combined with prior knowledge, researchers have used the genes Gfap, Aldh1I1, Slc1a3, and Gjb6 as markers for astrocytes(Srinivasan et al., 2016). Using the promoters for these genes,one is able to selectively express proteins in astrocytes that allow for the monitoring and manipulation of astrocytes. In particular relation to neurodegenerative intervention, viral strategies are emerging as a clinical candidate for targeting the CNS (Oswald et al., 2017).
Viral constructs targeting astrocyte gene expression
Viruses have been widely adopted as a way to introduce transgenes to the CNS for both preclinical and clinical studies. Compared to other methods such as nanoparticle delivery, viruses have been capitalized for their efficiency of gene transduction and long-term expression (Bouard et al., 2009).Viruses have been widely used to discover both basic and disease-oriented mechanisms regarding neurons, astrocytes,and microglia (Jahn et al., 2015; Luo et al., 2018). Inducing the expression of optogenetic and activity-based fluorescent proteins has allowed many research groups to both manipulate and monitor cells of the CNS in behaving animals. At the same time, clinical studies have attempted to use viruses to ameliorate deficits in neurological disease by modulating molecular or physiological deficits. Thus, we will review how viruses have been used to investigate astrocytes keeping in mind how these tools may be used to target astrocytes in clinical disease.
Researchers have used viruses to introduce genes implicated in neurodegenerative disease specifically into astrocytes in order to isolate astrocyte contributions to such diseases.There are a variety of viruses that have demonstrated tropism for astrocytes in the CNS. Lentiviruses, adeno-associated viruses (AAV), and canine adenovirus (CAV) have been successfully used to introduce transgenes to astrocytes.Combined with the ability to introduce astrocyte-promoter-dependent genes through viruses, a high astrocyte specificity can be achieved. While the majority of viral CNS studies have focused on neurons, recent studies demonstrate that such viral-based approaches are useful for understanding astrocyte function as well. For instance, using viral transduction strategies, astrocyte’s have been implicated in neurovascular coupling (Stobart et al., 2018), fear behavior(Martin-Fernandez et al., 2017), and feeding behavior (Chen et al., 2016) among other aspects of brain function.
Lentiviruses have been used successfully to target astrocytes in mice, and offer several advantages (Colin et al., 2009;Faideau et al., 2010; Delzor et al., 2013; Fassler et al., 2013).Lentiviral transgenes are transduced with high efficiency,with long term expression that is often required for in the context of neurodegenerative models (Park, 2007). Using a lentiviral strategy, Faideau et al., 2010 revealed that astrocyte expression of mutant Htt contributes to the mouse Huntington disease phenotypes. Aside from this study, Fassler and colleagues have developed a lentivirus construct that has preferential tropism for astrocytes (Fassler et al., 2013). Their construct expresses an anti-GLAST IgG on the lentivirus’surface. GLAST is an amino acid transporter linked to astrocyte synapse support function and is highly expressed in astrocytes (Zhou et al., 2006), thus, preferentially targeting astrocytes
In comparison to lentiviruses, AAVs have been much more commonly used to study astrocytes in both health and disease. Several serotypes of AAVs (AAV2/1, AAV2/5,AAV2/8, AAV2/9) have been tested to have tropism for astrocytes (Foust et al., 2009; Tong et al., 2014; Hammond et al., 2017; Pignataro et al., 2017). Using AAV transduction(serotype 2/5), the Khakh group was able to show that astrocyte expression of the potassium channel Kir4.1 rescues deficits in a Huntington disease mouse model (Tong et al.,2014). This study demonstrated that astrocytes play a role in regulating the excitability of neurons in the dorsal striatum,the principle region implicated in Huntington disease. Further demonstrating the role of astrocytes in Huntington disease, Meunier et al. (2016) have also used the AAV2/5 vector to selectively express mutant forms of Huntingtin in striatal astrocytes. They demonstrated that astrocyte expression of mutant Huntingtin contributes to pathological features of the Huntington mouse model. In comparison to AAV2/5 serotypes, researchers have demonstrated that AAV2/8 may transduce astrocytes with the higher efficiency in the CNS(Aschauer et al., 2013). Martin and colleagues used the AAV2/8 vector to employ a chemogenetic strategy to manipulate astrocytes. This work demonstrated the role astrocytes play in the central amygdala for fear and anxiety behaviors.Overall, AAVs represent a feasible tool to manipulate the genes of astrocytes and will likely continue to facilitate our understanding of astrocytes in a variety of disease states.
Our laboratory has recently reported that intravenously delivered CAV2 targets perivascular astrocytes throughout the adult brain (Kirschen et al., 2018). This has translational implications as other methods described above for transgene introduction is invasively hindering. Additionally,perivascular astrocytes in particular have been implicated in neurodegenerative disease (Montagne et al., 2015). Another advantage of this viral delivery strategy is that it can systemically manipulate astrocytes. Methods of stereotactic infusion may be region-specific, but it would not allow for the analysis of astrocytes in multiple communicating brain regions.Stereotactic infusion is also well known to cause reactive astrogliosis and thus may confound studies of astrocyte-intrinsic behavior (Dashkoff et al., 2016). Overall this finding indicates that CAV2 may provide tool to investigate the role of astrocytes in disease.
In the clinical realm, gene therapy via viral transduction has emerged as a promising method for addressing diseases of the CNS (Hocquemiller et al., 2016). There are varieties of ongoing clinical trials using intraparenchymal infusion of viruses in order to treat neurodegenerative diseases by modulating gene expression. For instance, intrastriatal infusion of AAVs has been used in phase I/II clinical trials for PD(Marks et al., 2008, 2010; Christine et al., 2009). While these represent landmark efforts for neurological therapy, these viruses introduced genes under the control of ubiquitous promoters and thus may induce gene expression in neurons and astrocytes alike. As mentioned previously, a major utility of genetic strategies is the ability to target specific cell types implicated in disease. In order to consider how astrocytes may be targeted in disease, it will be important to consider what promoter elements may be more specific to astrocytes(Cahoy et al., 2008). We list astrocyte-specific promoters(Gfap, Slc1a3, and Gjb6) used previously (Dabir et al., 2006;Nomura et al., 2010; Magnusson et al., 2014; Aida et al.,2015; Benedykcinska et al., 2016), with Aldh1I1 (Chai et al., 2017; Octeau et al., 2018; Yu et al., 2018) being recently validated as the most specific astrocyte promoter in murine studies (Srinivasan et al., 2016). One major caveat of using common promoters of astrocytes is the difficulty for packaging promoters of all sizes into viral vectors. For instance, it has been reported that AAV vectors have a limited physical space that may be required for large mammalian promoters(Dong et al., 1996; de Leeuw et al., 2016). To circumvent this issue, the Simpson group has worked on creating a variety of shortened promoters, termed “MiniPromoters” in order to target specific cell populations of the brain (de Leeuw et al.,2016). This work will be essential to identifying constructs that will allow gene manipulation specifically in astrocytes of patient brains.
Transgenic mouse models for studying astrocytes in neurodegeneration
Transgenic mouse models are widely used to study astrocytes. On the other hand, in the context of neurodegenerative disease, there has traditionally been a neuron-centric bias in which neuronal protein expression is thought to be the principal if not exclusive link to pathogenesis. Transgenic lines have allowed for the determination of glial cells’specific contributions to disease. For instance, researchers have used of the Cre/loxP system to restrict gene expression to astrocytes. Astrocyte promoters of Gfap and Ald1I1 have been used to express Cre recombinase in astrocytes (Jahn et al., 2015; Srinivasan et al., 2015, 2016). When introducing Cre-dependent genes, either through a double-transgenic cross or through viral transduction, one is able to manipulate genes specifically in astrocytes. Using a Cre-based conditional deletion scheme, the Cleveland lab showed that astrocytes play a major role in the progression of amyotrophic lateral sclerosis in the mutant SOD1 mouse model (Yamanaka et al.,2008). Researchers have used similar transgenic mouse line approaches to selectively express genes in astrocytes in the contexts of Parkinson disease, Alzheimer disease, and Huntington disease (Bradford et al., 2009; Gu et al., 2010; Lian et al., 2016; Wood et al., 2019). These studies served to dissect the contribution of astrocytes to neurodegenerative models.Recently, the Khakh lab generated an Aldh1I1 Cre line that allows for the specific expression of genes in all astrocytes of the CNS (Srinivasan et al., 2016). As opposed to previous lines based on other promoters such as GFAP and GLAST,these mice serve to restrict genetic expression to be more highly specific for a wider range of astrocytes in the CNS.This will allow for more sophisticated experiments in exploring the specific contribution of astrocytes to disease states.
Off-target effects of genetic strategies
Basic neurobiologists and developers of pharmaceuticals alike are wary of potential off-target manipulations that accompany both viral manipulation and transgenic mouse development. In terms of using viral constructs to target astrocytes in clinical contexts, immunogenic responses and oncogenic effects are principal concerns (Cotter and Muruve,2005; Marks et al., 2008). As illustrated above, there has been an immense interest in using AAVs for therapeutic intervention for their relatively non-immunogenic and non-oncogenic profiles (Hocquemiller et al., 2016; Naso et al., 2017).However, it should be noted that patients can develop antibodies against AAVs and give rise to the risk of developing immune responses to AAVs (Louis et al., 2013). Fortunately,strategies that locally target CNS regions (intraparenchymal)benefit from being relatively immune privileged systems(Christine et al., 2009). In contrast, recent preclinical studies indicate that systemic introduction of high-dose AAVs may result in severe toxicity affecting the nervous and hepatic systems (Hinderer et al., 2018; Hordeaux et al., 2018). While local CNS delivery of AAVs appear to be clinically promising, mechanisms of intracellular transport and transynaptic spread in the CNS remains unclear (Nonnenmacher and Weber, 2012; Castle et al., 2014; Zingg et al., 2017). Thus, the specificity of CNS viral delivery may be difficult to ascertain,and future studies are needed to determine whether or not astrocytes may be targeted in a region-specific manner. Lastly, one potential way to more specifically target astrocytes is to utilize astrocyte promoters studied in transgenic models in order restrict gene expression to astrocytes. However, there are two problems with this strategy. As discussed above,astrocytes display heterogeneity and traditional promoters such as GFAP and GLAST may be neither astrocyte-specific nor pan-astrocyte targeting throughout the CNS (Sofroniew,2009; Srinivasan et al., 2016). In addition, recent studies have remarked that there are significant differences between murine and human astrocytes, potentially confounding the contribution of astrocyte-specific genes and pathology to neurological diseases (Vasile et al., 2017). Together, these points serve as cautions for considering astrocyte gene therapy and for interpreting studies of astrocytes in general.
Conclusions and Perspective
Based on a wealth of recent investigations, the specific roles and molecular pathways implicated in astrocytes’ response to neurodegenerative injury are becoming increasingly clear.For instance, while it is traditionally held that astrocytes interact principally if not exclusively with neurons, the complex cross-talk between astrocytes and microglia especially during injury has been acknowledged and is the subject of much clinical interest. Based on the existing viral and genetic tools in the field, a number of important astrocyte-autonomous functions in neurodegenerative diseases have been uncovered, and likely more remain to be seen. Exciting new work is beginning to profile the astrocyte inflammasome in various pro-inflammatory settings (Dozio et al., 2018). The better these functions are understood, the better they can be counteracted or modulated, with the goal of controlling neuroinflammation and promoting reparative physiology rather than exacerbating injury and further compounding neurodegenerative phenotypes. Ultimately, much of this work still rests on mammalian model systems, and our genetic understanding of astrocytes, although emerging technology may allow monitoring astrocyte activity in humans (Edison et al.,2018). These emerging technologies, in tandem with clinical genetic strategies, may lead to targeted treatment of astrocyte-mediated defects in neurodegenerative disease. Overall,there is likely a lot of untapped power in astrocytes that may prove fruitful for future clinical studies in the context of currently unmodifiable neurodegenerative diseases.
Author contributions:All authors wrote the manuscript and approved the final manuscript.
Conflicts of interest:authors declare no conflicts of interest.
Financial support:The work was supported by National Institutes of Health (NIH) Grants 1F30MH110103 (to GWK) and 1F30MH116650 (to RK).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by
Peer review:Externally peer reviewed.
Open access statement:is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Ethanol extract from Gynostemma pentaphyllum ameliorates dopaminergic neuronal cell death in transgenic mice expressing mutant A53T human alpha-synuclein
- Peripheral nerve injury induced changes in the spinal cord and strategies to counteract/enhance the changes to promote nerve regeneration
- Pathological significance of tRNA-derived small RNAs in neurological disorders
- Applications of advanced signal processing and machine learning in the neonatal hypoxic-ischemic electroencephalography
- Protective effect of hydrogen sulfide on oxidative stress-induced neurodegenerative diseases
- Current status and future prospects of stem cell therapy in Alzheimer's disease