
HPLC 法测定(E)-4-[2-(4-氯苯氧基)-2-甲基丙酰氧基]-3-甲氧基苯基丙烯酸的有关物质
2020-10-29 09:25熊贤招
中国药科大学学报 2020年5期
许 静,彭 红,熊贤招
(江西中医药大学,南昌330004)
(E)-4-[2-(4-氯苯氧基)-2-甲基丙酰氧基]-3-甲氧基苯基丙烯酸((E)-4-[2-(4-chlorophenoxy)-2-methylpanoyloxy] -3-methoxyphenylacrylic acid,XTAC)是由江西中医药大学药物化学教研室合成的一种全新结构的降脂药物,药理学实验结果表明,XTAC能够显著降低血清中总胆固醇、总三酰甘油和低密度脂蛋白胆固醇,并可降低动脉粥样硬化指数[1],XTAC的合成工艺简单(共3步),合成途径如图1所示,前期已经研究了XTAC原料药中的残留溶剂[2],但是关于其他杂质的研究甚少,根据杂质研究指导原则及ICH杂质研究的要求,对其有关物质进行研究,以合成起始原料、中间体及可能产生的副产物或降解产物为研究对象,分析了XTAC产生的有关物质并建立了HPLC分析方法[3-4],分离并鉴定了主要杂质C[5],为 XTAC的质量控制提供了理论依据。
1 材料
1.1 仪器
e2695高效液相色谱仪(美国沃特世公司);Triple TOF 5600+型四极杆飞行时间串联质谱仪(美国AB SCIEX公司);AvanceⅢHD 600 MHz型核磁共振波谱仪(瑞士Bruker科技有限公司)。
1.2 试药
XTAC( 批 号 :20190324、20190401、20190412)、中间体2(批号:20190312)由江西中医药大学药物化学教研室提供;杂质C(批号:20190513)由本实验室自制。
2 方法与结果
2.1 HPLC色谱条件
色谱柱:Ultimate XB-C18色谱柱(4.6 mm×150 mm,5 μm);流动相:甲醇-1%乙酸水溶液(70∶30),等度洗脱;柱温:25 ℃;流速:1.0 mL/min;检测波长:275 nm;进样量:10 μL。
2.2 系统适用性
精密称取研细后的XTAC 100 mg,置50 mL量瓶中,加流动相适量超声使溶解,放至室温后加流动相稀释至刻度,摇匀,过滤,作为供试品溶液,照“2.1”项下色谱条件进行测定,记录色谱图(见图2)。理论塔板数按XTAC峰计大于2 500。
2.3 专属性
2.3.1 XTAC、起始原料以及中间体的分离度取一定浓度的本品、起始原料和工艺中间体的混合溶液,照“2.1”项下的色谱条件进行检测。结果见图3,XTAC与起始原料及中间体的分离度较好。
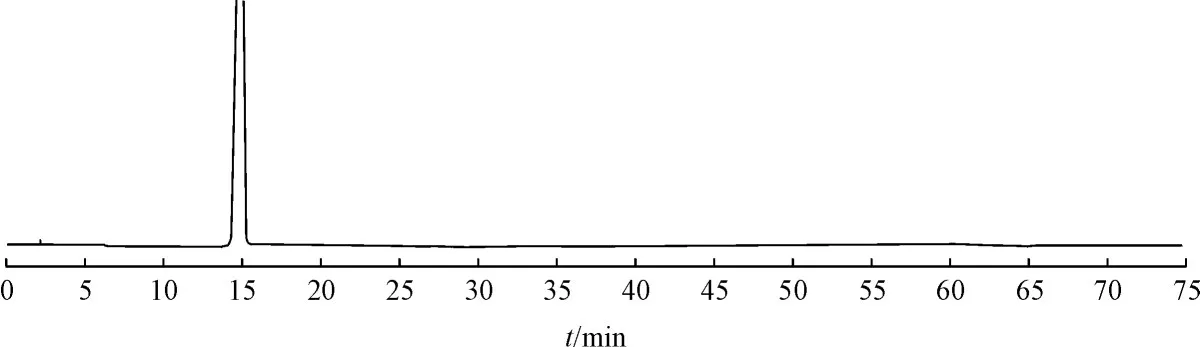
Figure 2 HPLC diagram of XTAC
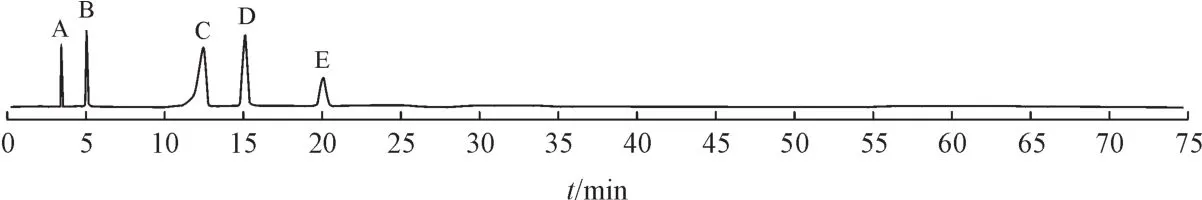
Figure 3 HPLC diagram of mixed solution of raw material(A),intermediate1(B),intermediate 2(C),XTAC(D)and impurity of intermediate 2(E)
2.3.2 强制降解实验 分别取XTAC约100 mg,精密称定,置不同50 mL量瓶中,进行强降解实验。未破坏供试品溶液:加流动相适量超声溶解;高温破坏供试品溶液:在105℃下恒温条件放置4 h,冷却至室温;光破坏供试品溶液:紫外光(4 500±500)lx下照射4 h,加流动相适量超声溶解;碱破坏供试品溶液:加1 mol/L NaOH溶液1 mL,在80℃水浴条件下加热30 min,放冷至室温后加1 mol/L的盐酸溶液中和至中性,加流动相适量超声溶解;酸破坏供试品溶液:加1 mol/L的盐酸溶液1 mL,在80℃水浴条件下加热30 min,放冷至室温后加1 mol/L的NaOH溶液中和至中性,加流动相适量超声溶解;氧化破坏供试品溶液:加30%H2O2溶液1 mL,在室温下放置30 min,加流动相适量超声溶解。上述样品用流动相稀释至刻度,摇匀,过滤,按“2.1”项下色谱条件进行检测,分离度符合要求,结果见表1。碱破坏、氧化破坏实验的色谱图见图4,图5。

Table 1 Forced degradation test results of XTAC

Figure 4 HPLC diagram of alkaline-treated sample

Figure 5 HPLC diagram of oxygen-treated sample
强制降解试验结果显示各降解产物均在主峰保留时间的2倍内出峰,为了继续考察本品在稳定性试验期间的有关物质,故确定本品有关物质检查保留时间为主峰保留时间的4倍。强制降解色谱图中主峰与各杂质峰分离度均大于1.5,各杂质间分离度良好,物料平衡在98.76%~100.67%之间符合验证要求。本品在酸、光照、高温、氧化条件下主成分含量均大于99.00%。本品碱条件较不稳定,主成分下降,杂质峰升高。未破坏、酸破坏、光照破坏、氧破坏样品中杂质C的含量最多。
2.4 主要杂质C的结构鉴定
2.4.1 样品中有关物质分析 3批XTAC样品纯度较高,有关物质含量均小于0.1%,含量较大的有关物质为杂质C,采用制备HPLC收集杂质C,HPLC图谱显示杂质C的保留时间与中间体2保留时间一致(图6)。

Figure 6 HPLC diagram of impurity C reference substance
2.4.2 杂质C的结构确证 杂质C为白色至类白色晶体,ESI-MSm/z349[M+H]+。1H NMR谱(600 MHz,DMSO-d6)如图7所示:δ9.98(1H,S,C16-H)、δ7.69(1H,s,C11、C2、C4、C5-H中的 3个)、δ7.38(6H,s,C1、C2、C4、C5、C10、C13-H)、δ3.87(1H,s,C15-H中的 1个)、δ1.61(6H,s,C18和C19-CH3)。13C NMR 谱(151 MHz,DMSO-d6)如图 8所示:δ192.47(C16);δ171.41(C14);δ154.06(C6)、δ151.88(C8)、δ144.15(C9)、δ135.90(C12)、δ129.64(C2 或 C4)、δ126.57(C3)、δ124.04(C1 或C5)、δ123.98(C1 或 C5)、δ123.81(C10)、δ121.12(C11)、δ112.50(C13);δ79.44(C7)、δ56.61(C15)、δ25.47(C17或C18)。结合总离子流图和一级、二级质谱图(图7,图8)可知,检测到m/z349.183 2的[M+H]+准分子离子峰,可知该物质相对分子质量为 348.5,tR为 9.51 min,在 MS/MS图上出现m/z111.001 2、m/z169.042 4的特征离子碎片,结合杂质C母核结构特征可知m/z169.042 4可能为杂质C的酯键进行α-断裂然后失去一分子中性分子CO生成的碎片,m/z111.001 2可能为杂质C的醚键进行i-断裂生成的碎片,同时利用同位素峰形匹配校正技术查找以及质谱碎片推测该物质分子式为C18H17O5Cl。

Figure 7 Primary mass spectrum of impurity C

Figure 8 Secondary mass pectrum of impurity C
2.5 杂质C的线性范围与检测限
取杂质C对照品约50 mg,精密称定,置100 mL量瓶中,加流动相超声溶解并稀释至刻度,摇匀,作为杂质C对照品溶液。精密量取杂质C对照品溶液2 mL,置10 mL量瓶中,加流动相稀释至刻度,作为杂质C对照品储备液。分别精密量取杂质C对照品储备液各0.02、0.1、1、2、4、6 mL,置10 mL量瓶中,加流动相稀释至刻度,制成系列浓度的杂质C标准溶液。照“2.1”项下色谱条件进行测定,记录色谱图。以质量浓度为横坐标,以峰面积为纵坐标,进行回归得回归方程:y=3650.6x+549.48(r=0.999 9),在 0.20~59.96 μg/mL范围内杂质C的质量浓度与色谱峰面积呈良好的线性关系。精密量取杂质C对照品储备液逐级稀释至信噪比(S/N)约为3时,检测限为15 ng。
2.6 加样回收率
XTAC研细,取约50 mg,精密称定,置100 mL量瓶中,加流动相50 mL超声使溶解,加杂质C对照品溶液(8 μg/mL)2.0,2.5,3.0 mL(各3份),再用流动相稀释至刻度,摇匀,作为供试品溶液;精密称取杂质C(中间体2)对照品约5 mg,置100 mL量瓶中,加流动相适量超声使溶解,用流动相稀释至刻度,摇匀,精密量取杂质C对照品储备液4 mL,置25 mL量瓶中,加流动相稀释至刻度,摇匀,作为杂质C对照品溶液。照“2.1”项下色谱条件测定含量,平均回收率为92.88%,RSD为5.62%。结果见表2。
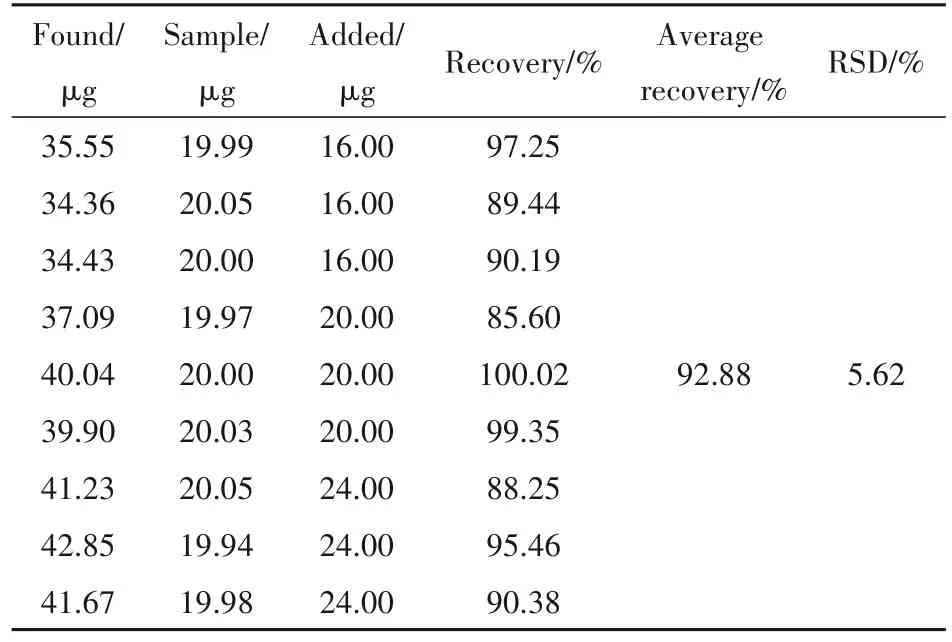
Table 2 Accuracy test results of impurity C
2.7 有关物质检测
取XTAC样品细粉适量(约相当于XTAC 50 mg),精密称定,置50 mL量瓶中,加流动相适量超声溶解,加流动相稀释至刻度,摇匀,过滤,作为XTAC供试品溶液;精密量取XTAC供试品溶液1 mL,置100 mL量瓶中,加流动相稀释至刻度,摇匀,作为XTAC对照溶液。另取杂质C(中间体2)对照品适量,精密称定,用流动相超声溶解并定量稀释成每毫升含40 μg的溶液,作为杂质C对照品溶液。按“2.1”项色谱条件进行测定,记录色谱图至供试品溶液主峰保留时间的4倍。理论板数按XTAC对照溶液峰计算应不低于2 500,用XTAC对照溶液的色谱图来确定杂质C,杂质C相对于XTAC(保留时间大约为15.6 min)的相对保留时间约为0.82。除溶剂峰外,XTAC供试品溶液色谱图中如有与对照品溶液主峰保留时间一致的色谱峰,按外标法以峰面积计算不得过1.0%;XTAC供试品溶液色谱图中如有杂质峰,单个杂质峰面积不得大于XTAC对照溶液主峰面积的0.5倍(0.5%);各杂质峰面积的和不得大于XTAC对照溶液主峰面积的2倍(2.0%)。供试品溶液色谱图中小于主峰面积的0.005倍色谱峰忽略不计(0.005%)。3批XTAC样品的有关物质检测结果见表3。结果显示,XTAC样品的纯度较高,杂质的表观含量均在0.1%以下。

Table 3 Determination of related substances in XTAC
3 讨 论
本研究对XTAC样品中主要有关物质C进行了初步结构确证,并拟定了杂质C对照品的质量要求,在XTAC原料药质量标准中采用杂质对照品外标法控制样品中杂质C的限量;对其他的0.1%以下杂质,采用高低浓度对比法进行单个杂质限量控制,并对总杂质限量进行了控制,通过分析方法验证,表明该方法适用于本品的有关物质分析,在后续经稳定性、放大生产及进一步的药理毒理研究,在获得更多的信息下,将对这些有关物质会进一步进行评价和质量控制研究。本研究所建立的HPLC方法简单、方便、稳定,能够有效地进行XTAC有关物质的测定。
根据欧盟发布的警示结构,XTAC没有基因毒性杂质警示结构,但杂质C和其他杂质3-甲氧基对羟基苯甲醛具有基因毒性杂质警示结构(SA_11:Simple aldehyde,)。虽然杂质C的含量小于0.1%(约0.05%),其他杂质含量也远远小于杂质C的含量,但其相关基因毒性杂质的研究仍应得到重视。
猜你喜欢
佳木斯大学学报(自然科学版)(2022年1期)2022-11-25
中国兽药杂志(2022年6期)2022-07-04
中国药学药品知识仓库(2022年13期)2022-07-03
中国药学药品知识仓库(2022年10期)2022-05-29
药品评价(2021年17期)2021-11-06
汕头大学学报(自然科学版)(2020年4期)2020-12-14
学苑创造·A版(2019年9期)2019-11-07
学苑创造·B版(2017年1期)2017-02-21
学苑创造·B版(2017年1期)2017-02-21
小天使·二年级语数英综合(2016年9期)2016-05-14
