
基于ddGBS的桉树SNP挖掘和系统进化分析
2020-11-03 07:55贾翠蓉朱显亮周长品翁启杰甘四明李发根
中南林业科技大学学报 2020年10期
贾翠蓉,朱显亮,王 莉,周长品,翁启杰,甘四明,李发根
(1.中国林业科学研究院 a.热带林业研究所;b.热带林业研究国家林业和草原局重点实验室,广东 广州 510520;2.南京林业大学 林学院,江苏 南京 210037)
桉树隶属于桃金娘科Myrtaceae,主要包括三大属:桉属Eucalyptus、杯果木属Angophora和伞房属Corymbia,主要分布于澳大利亚,有808 个种和137 个变种[1]。大部分桉树具有雄蕊先熟、花期不遇和地理隔离的特征,这使其自交或近交概率极低,但在重叠邻接的桉树自然分布区或引种栽培区有可能产生自然或人工杂种[2-4]。桉树经过世代的遗传变异和自然选择,逐步演化出种类庞杂的桉树体系[5],因此,需要借助分子标记的手段来研究其系统进化历程。
单核苷酸多态性(Single nucleotide polymorphism, SNP)由于其在基因组上分布广泛、易于高通量检测和分型等优点广泛应用于资源评价和遗传图谱构建等研究[6-7]。随着高通量测序技术的发展,测序成本大幅降低。对于群体遗传研究而言,仅需要分布均匀且一定数量的SNP 标记即可,基于测序的基因分型(Genotyping by sequencing,GBS)技术成本低、操作简便和高通量等特点使其成为SNP 挖掘的有效方法[8]。ddGBS(Double digest GBS)主要利用双酶切降低基因组的复杂度,使用标签序列区别样本,实现了大量样本的测序分型[9],广泛应用于种质资源的遗传结构和系统进化分析[10-11]。ddGBS 技术在有或无参考基因组的情况下,均可通过对测序结果进行拼接组装,开发度SNP 标记[12-13]。
多种分子标记技术已应用于桉树群体遗传和系统进化研究,如随机扩增多态性(Random amplified polymorphic DNA, RAPD)、简单重复序列(Simple sequence repeat, SSR)和扩增片段长度多态性(Amplified fragment length polymorphism,AFLP)标记以及多样性阵列技术(Diversity array technology, DArT)[14-18]。尽管利用分子标记对桉树的研究已开展多年,但仍有许多系统发育问题和分类问题尚待解决。而准确的系统发育对于理解进化具有重要的作用,应用也非常广泛。例如将进化多样性纳入生物多样性测量指标[19],为生态学研究提供系统发育观点[20],预测物种对害虫和病原体的敏感性[21]等。本研究利用ddGBS 技术对14 种不同桉树(28 份样品)进行建库测序、开发高质量SNP 标记并进行基因分型,通过开展系统进化树的构建研究,为桉树的分类、分子鉴定及种质资源评价提供大量的标记资源。
1 材料与方法
1.1 试验材料
本试验用的14 种桉树来自课题前期收集的材料,分别是:柠檬桉C.citriodora、方格皮桉C.tessellaris、粗皮桉E.pellita、尾叶桉E.urophylla、亮果桉E.nitens、邓恩桉E.dunnii、蓝 桉E.globulus、赤 桉E.camaldulensis、细 叶桉E.tereticornis、加拉桉E.marginata、银顶山岑桉E.sieberi、克蒂斯桉E.curtisii、四齿桉E.tetrodonta和王桉E.regnans。每种桉树选择2 棵,均来自不同的家系(详细的种源信息及分类学状态列于表1)。物种名称和分类学来自Hill 和Johnson 于1995年公布的分类系统[1]。
1.1.1 DNA 样品制备
采用改良的CTAB 法提取基因组DNA[22],保存于-80℃冰箱备用,使用琼脂糖凝胶电泳检测DNA 质量,并利用超微量分光光度计(Nanodrop 2000, Thermo fisher scientific)测定DNA 浓度和纯度(即OD260 与OD280 的比值分布在1.8~2.0之间),再利用Qubit 3.0 荧光定量仪(Thermo Fisher scientific)对样品DNA 进行定量。
1.1.2 GBS 方案选择及测序文库构建
建库主要参照Poland 等[23]方案,选取在基因组上酶切位点分布较多的MspI 酶(C|CGG)和分布较少的PstI 内切酶(CTGCA|G)以减少基因组的复杂性。首先利用Restriction Digest 酶切软件(https://github.com/JINPENG-WANG/RestrictionDigest)分析巨桉参考基因组序列(https://phytozome.jgi.doe.gov),预估MspI 和PstI 的 电子酶切效果,推测其在目标文库范围内的酶切片段数。参照Poland 等[23]的建库流程(酶切—连接—混样—PCR 扩增—纯化),本研究将混样环节改进为:等纳摩尔质量混样,双轮磁珠分选350 bp 片段,分选磁珠采用诺唯赞VAHTS DNA Clean Beads。使 用Thermo Fisher Scientific Qubit 3.0 检测文库浓度,之后利用PekinElmer Labchip GXII Touch 微流控毛细管电泳系统对文库进行质检。质检合格后送至北京贝瑞基因进行双端测序(PE 150),测序平台为Illumina HiSeq X Ten。
1.2 数据分析
1.2.1 SNP 开发与注释
利用Stacks V2.41(http://catchenlab.life.illinois.edu/stacks/)软件拆分下机数据,按照如下标准去除低质量序列:序列中N 的比例≥10%及序列中>50%的碱基质量低于5 的序列。去除低质量序列后通过Bowtie2 v2.2.2.9(https://github.com/BenLangmead/bowtie2)软件与巨桉基因组进行比对,然后利用GATK 3.8(https://software.broadinstitute.org/gatk/)软件开发SNP 位点。
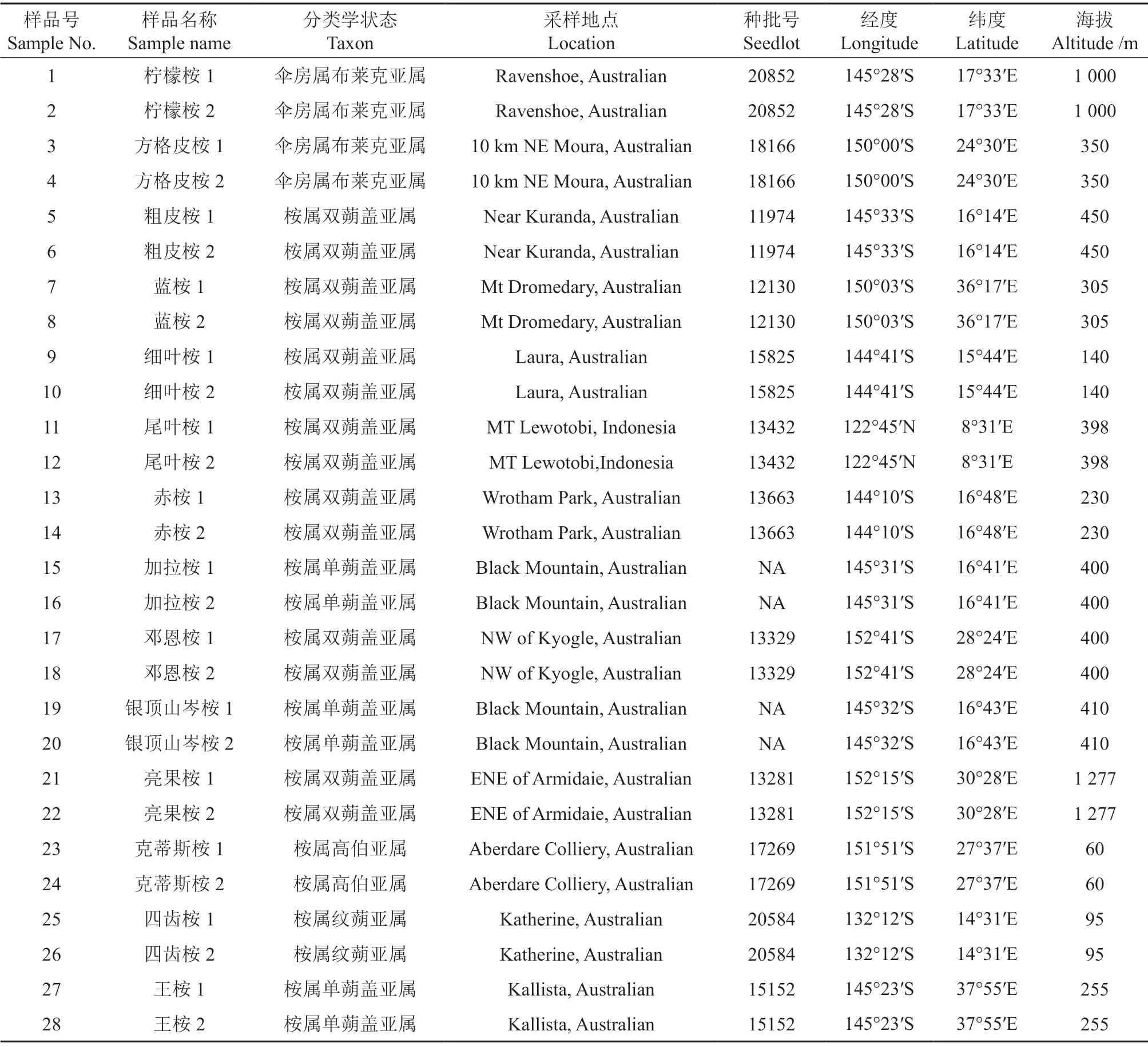
表1 14 种桉树的分类学状态及种源家系信息†Table 1 The information of fourteen Eucalyptus used for analysis
使用Vcftools v4.2(http://vcftools.sourceforge.net/)软件对检测到的SNP 位点数据进行了过滤,过滤参数为:最小平均测序深度为4,无缺失数据,最小杂合比为0.05,从而获得高质量的SNP。使用SnpEff v4.3(http://snpeff.sourceforge.net/)软件对这些SNP 位点进行注释。
1.2.2 系统进化树的构建及核苷酸多样性分析
基于过滤后的高质量SNP 位点,利用IQTREE v1.6.11(http://www.iqtree.org/) 软件采用最大似然法(Maximum Likelihood, ML) 对28个样品进行聚类分析,构建系统进化树,再利用Figtree v1.4.4(http://tree.bio.ed.ac.uk/software/figtree/)软件生成可视化树文件。
利用Vcftools 软件计算核苷酸多态性,以200 kb 为滑动窗口,100 kb 为步长,进行核苷酸多样性(两条序列中不同核苷酸的比例)分析。
2 结果与分析
2.1 测序质量及SNP 分布
基因组DNA 经双酶切建库测序,去除低质量序列后,共获得30.49 Gb 数据,平均每个样品的数据量为1.09 Gb。如表2所示:测序共产生210 276 036 条序列,样品平均序列条数为7 509 858,14 个树种产生的测序片段数存在差异,其中最多的为加拉桉,平均测序片段数为10 913 288 条,最少为蓝桉,平均产生4 741 137 条。测序Q30 值在97.7%~98.3%范围内,平均为98.0%;GC 含量为43%~47%,平均的GC 含量为44%。样品间比对匹配率在46.0%~88.9%之间,平均为77.8%,其中,比对匹配率最高为尾叶桉(88.9%),最低为方格皮桉(45.0%)。各树种与巨桉平均对比匹配率在47%~49%之间的是方格皮桉和柠檬桉;在66%~84.5%范围的包括加拉桉、银顶山岑桉、四齿桉、王桉和克蒂斯桉;在86%~89%范围的是粗皮桉、赤桉、蓝桉、细叶桉、尾叶桉、邓恩桉和亮果桉。
利用GATK 软件共检测到2 606 828 个SNP位点,经Vcftools 软件对SNP 位点数据进过滤后,保留下42 222 个高可信度SNP。根据绘制的SNP 位点在巨桉参考基因组上的分布(图1)可见,SNP 位点在染色体上分布较为均匀,约99.6%(42 059)的SNP 成功定位在巨桉参考基因组11条染色体上,仅有约0.4%(163 个)的SNP 无法定位到11 条染色体上。对高可信度SNP 进行功能注释,统计结果(表3)表明:SNP 标记约有29.0%位于外显子区域,内含子区、上游和下游区域分别有11.3%、13.2%和12.4%,5′和3′端的UTR 区含有5.1%的SNP 位点,剪切位点为2.2%,基因间区仅有1.9%。
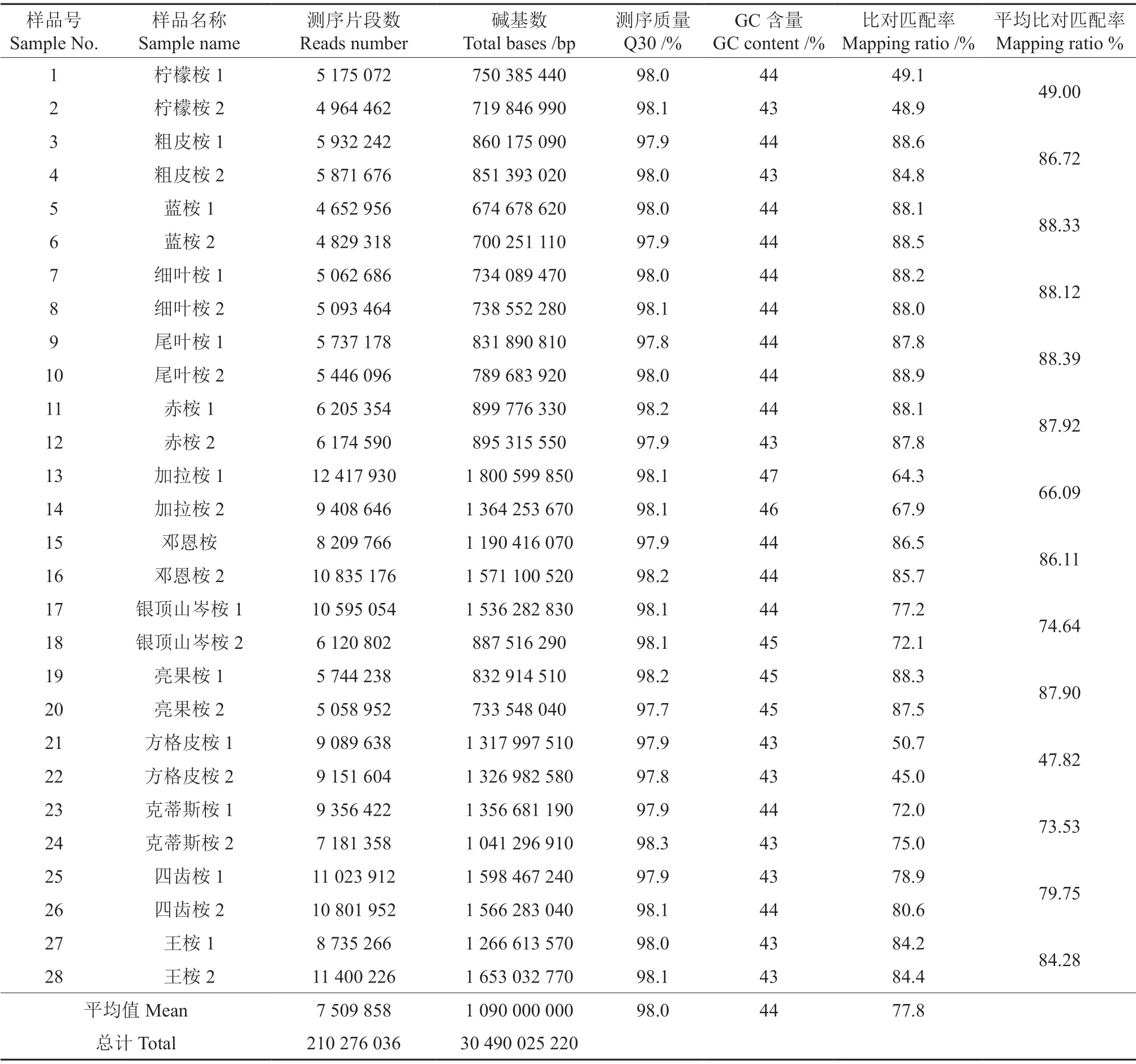
表2 桉树GBS 测序数据统计Table 2 GBS sequencing data statistics in fourteen eucalyptus
2.2 进化树构建及多样性分析
基于42 222 个高质量SNP,统计各树种的特有SNP 位点(即仅在该树种存在的SNP 位点)数目(表4),分别包括样品特有SNP 位点数目和物种特有SNP 位点数目。14 种桉树的SNP 数目存在差异,其中克蒂斯桉(3 348 个)、方格皮桉(1 509 个)和王桉(1 610 个)的特有SNP 较多,柠檬桉(841 个)、加拉桉(593 个)次之,粗皮桉(57个)和细叶桉(56 个)最少。14 种桉树的核苷酸多样性值主要分布在1.03×10-6~1.07×10-4之间,最大值为1.95×10-4,平均值为2.82×10-5(图2)。

图1 SNP 位点在巨桉基因组染色体上的分布Fig.1 Distribution of SNP loci on the chromosome of E.grandis genome

表3 SNP 注释结果Table 3 The results of SNP annotation

表4 桉树树种的特有SNP 位点统计†Table 4 The statistics of unique SNP in fourteen eucalypts

图2 14 种桉树的核酸多样性Fig.2 Nucleotide diversity value of 14 species in eucalypts
ML 法构建的进化树结果(图3)显示:14 个树种可以聚为2 大类,聚类一是伞房属布莱克亚属的柠檬桉和方格皮桉,聚类二是其他12 个树种。聚类二分为2 个分支:第一分支包括单蒴盖亚属的加拉桉、王桉和银顶山岑桉,高伯亚属的克蒂斯桉、纹蒴亚属的四齿桉。第二分支含有7 个来自双蒴盖亚属的树种:粗皮桉、蓝桉、细叶桉、尾叶桉、赤桉、亮果桉以及邓恩桉,其中粗皮桉和尾叶桉同属横脉组脂桉系,细叶桉和赤桉隶属于窿缘组,邓恩桉、亮果桉和蓝桉同属蓝桉组多枝桉系。

图3 14 种桉树的系统进化树Fig.3 Systematic evolutionary tree of fourteen species in eucalypts
3 结论与讨论
本研究利用ddGBS 技术对14 个桉树树种进行测序分型,发掘42 222 个高质量SNP 位点,可广泛应用于桉树SNP 开发。并在此基础上进行系统进化树分析,结果与Hill 和Johnson 的桉属双蒴盖亚属及伞房属的分类结果一致,同时也和新的分类系统保持一致,这为桉树系统分类提供新思路,对于进一步开展桉树关联遗传学研究和重要性状QTL 定位等方面具有重要的应用价值。
GBS 技术受内切酶种类的限制,有研究表明物种酶切效率因选择的内切酶不同而有所差异[24],良好的酶切组合有利于提高文库质量,增加标签数量和SNP 位点数[25]。目前在植物中,ApeKI、PstI 和EcoRI 通常与MspI、MseI 和HpaII 等酶结合使用,这取决于每个物种的基因组特异性。前期陈升侃等[26]利用EcoRI 和HindIII 酶切组合开发尾叶桉×细叶桉杂种SNP 位点,并应用于材性性状的关联分析;Timothy 等人[27]利用GBS 技术和桉树60K 芯片对蓝桉无性系子代群体进行SNP开发及分型,比较发现GBS 跟桉树60K 芯片有13 645 个不同的SNP 位点;Collins 等[31]采用单酶PstI 从基因组学的角度证明E.magnificata是一个独特的分类群。本研究采用PstI 和MspI 内切酶组合来开发桉树高质量SNP 标记,并应用于桉树系统发育研究。
形态学、植物化学和基因组学分析均在桉树分类研究中发挥重要作用[28-30],其中基因组数据是阐明物种进化和分化过程中的有力工具。二代测序数据在很大程度上与形态学和植物化学分析一致,同时也能进一步推断出不同种群之间的变异[31]。基于ddGBS 技术的进化树构建为桉树系统发育研究提供了良好且低成本的技术支持。如Collins 等[31]整合形态学、植物化学和GBS 的方法发现并鉴定了一个桉树新的稀有种,并且GBS的结果也与形态学和植物化学的结果相互印证。前人的研究表明,柠檬桉和方格皮桉同属于伞房属的布莱克亚属[1],本研究构建的系统发育树中,伞房亚属的柠檬桉和方格皮桉聚为一类,这也完全与桉树的最新版分类系统一致,证实了本研究的可靠性[32]。
前期Steane 等[16]和Jones 等[17]利用可覆盖全基因组的DArT 标记对双蒴盖亚属桉树进行了大样本多类群的系统发育研究,为进化和生态研究提供了一个强有力的系统发育框架。Nicolle 等人[33]也在Jones 和Steane 研究的基础上加入野外调查数据、植物标本及其他系统发育的研究结果,对双蒴盖亚属桉树分类进行了补充和修订。本研究采用的ddGBS 技术以经济简便的方法开发SNP 来构建系统发育树,其中双蒴盖亚属中的树种聚为平行的三个分支:横脉组脂桉系(粗皮桉和尾叶桉)、窿缘组(细叶桉和赤桉)及蓝桉组多枝桉系(邓恩桉、亮果桉和蓝桉),结果与上述的研究基本一致[17,33],同时与Hill 等的分类高度一致。其他亚属的分类结果,如高伯亚属的克蒂斯桉、纹蒴亚属的四齿桉、单蒴盖亚属的加拉桉、王桉和银顶山岑桉,与Hill 等的分类系统及Nicolle 的最新分类系统一致性较高。
14 个桉树树种与巨桉参考基因组的平均比对匹配率存在差异,其分布规律与进化树存在相似性,故推测可能是缘于树种与巨桉亲缘关系远近不同有关,如来自桉属的粗皮桉、蓝桉、细叶桉、尾叶桉、赤桉、亮果桉以及邓恩桉与巨桉同属双蒴盖亚属,其比对匹配率较高(86%~89%),而亲缘关系较远的来自伞房属的柠檬桉和方格皮桉,其匹配率则低于50%。
GBS 技术以其操作过程简单且使用甲基化敏感酶回避了基因组重复区域成为了简化基因组测序的主流技术,当前也广泛应用于系统发育、全基因组关联、QTLs 定位、连锁图谱构建等领域[10-12]。本研究验证了ddGBS 方法构建进化树的可靠性,在后续的试验中,将通过使用更多桉树基因组特异性的酶切组合,以高效的开发桉树SNP 标记。Thornhill 等人[34]通过matK 和psbA-trnH 基因,及核内转录间隔区、外转录间隔区的DNA 序列,来评估桉树的分类与711 个物种的系统发育的匹配度,发现杯果木属不能单独成为一个属,而应归于伞房属,或者两个属亲缘关系非常近。下一步研究可增加其他更多桉树树种的分析,例如杯果木属,这对于探索杯果木属与伞房属的遗传进化关系具有重大的意义。
猜你喜欢
军事文摘(2022年14期)2022-08-26
农村科学实验(2021年27期)2021-11-12
天津市教科院学报(2021年5期)2021-11-10
儿童故事画报·发现号趣味百科(2019年9期)2019-02-02
环球时报(2019-01-03)2019-01-03
作文大王·中高年级(2018年5期)2018-06-20
作文大王·中高年级(2017年11期)2018-03-07
作文大王·中高年级(2017年9期)2017-09-22
中国医学创新(2017年7期)2017-03-31
江苏农业科学(2016年8期)2017-02-15
