
类水滑石材料主客体插层结构的构筑及特性的理论研究
2020-11-13 09:37朱玉荃赵晓婕陈子茹
高等学校化学学报 2020年11期
朱玉荃,赵晓婕,钟 嫄,陈子茹,鄢 红,段 雪
(北京化工大学化学学院,化工资源有效利用国家重点实验室,北京100029)
层状复合金属氢氧化物(Layered double hydroxides,LDHs)也被称作类水滑石,是一类典型的阴离子型主客体层状结构化合物.其主体层板中的金属离子与羟基形成六配位的八面体构型,八面体单元共边构成了带电荷的LDHs二维层板,这些层板沿第三维方向堆叠形成有序层状结构.同时客体阴离子进入层间以平衡层板正电荷[1].其基本的化学组成可表示为(图1),其中,MⅡ为二价金属阳离子,如Mg2+,Zn2+,Cu2+,Ni2+,Ca2+,Mn2+,Co2+和Fe2+等[1];MⅢ为三价金属阳离子,如Al3+,Ga3+,Fe3+,Cr3+,Mn3+和Co3+等[1]. 此外,Li+和Ti4+也有被引入主体层板的报道[1];An—为层间阴离子,常见的为简单的无机阴离子(Cl—,OH—和CO23-等)[1]、各种有机羧酸根(包括药物分子)[4~7]、配合物阴离子[8~12]、聚合物[13,14]及生物活性分子[15]等均已被引入层间.m表示结晶水的数量,MⅡ/MⅢ的比值(R)通常在2~4范围内[16].晶体学上,LDHs常见的堆积方式有六方(Hexagonal)2H和斜方(Rhombohedral)3R两种[17]. 由于其主客体结构高度可调,其在催化[18]、吸附与分离[19,20]、离子交换[21]、生物/医药[22]以及光、电、磁等功能材料[2,23~25]等领域表现出重要的应用价值,受到学术界和产业界的广泛关注.

Fig.1 Idealized structure of layered double hydroxides(LDHs)
通常,材料的性能取决于其结构,故LDHs的结构设计与构筑是其进一步应用的基础.但是,由于LDHs结构复杂不易获得单晶结构,虽然X射线光电子能谱(XPS)、X射线近边吸收光谱(XANES)、扩展X射线吸收精细结构谱(EXAFS)等一系列实验表征技术已成功地用于揭示某些LDHs的微观结构[26],但对其结构的认识仍存在很多未解决或有争议的问题,如,哪些离子能够引入层板、层板阳离子的MII/MIII比值(R)在什么范围内可以得到纯相LDHs、层板的电荷分布情况如何、层板阳离子的有序性、复杂阴离子在LDHs层间具体的排列方式、层板结构的堆积方式以及主客体相互作用的本质等.
近年来,随着计算技术的飞速发展,利用电子结构方法和分子模拟方法来研究LDHs材料的微观结构已成为继实验方法后的有力研究手段.在电子结构研究方法中:一种是基于团簇模型,利用从头算或密度泛函(DFT)方法,以小的原子簇代替层状材料的结构来研究层板结构的详细信息[27];另一种是采用周期性固体模型,利用基于DFT的平面波赝势方法(Plane wave density functional theory,PWDFT)和周期边界条件(Periodic boundary condition,PBC),用从头算方法求解固体的单电子Schrödinger方程或Kohn-Sham方程得到能带和波函数[28,29].这种方法既可计算晶体的微观结构,也避免了团簇模型不能进行整体性描述的缺点,是目前研究固体微观结构的一种非常有效的计算方法.另外,以力场为基础的分子动力学模拟方法(Molecular dynamics simulation)主要用来考查层间客体的性质及其与层板的相互作用[30].
本文系统综述了上述理论方法应用于LDHs结构构筑及特性研究的最新进展,包括主体结构、客体结构和主客体相互作用3个方面的研究工作进展,及其在作为光驱动催化剂方面应用的理论研究[31].揭示了LDHs材料结构-性能之间的构效关系,为以LDHs为材料平台构筑一系列基于其超分子插层结构主客体间相互作用的新型功能材料、扩展材料的功能性提供理论指导.
1 LDHs计算模型和计算方法
1.1 LDHs计算模型
LDHs计算模型通常有团簇模型和周期性固体模型两种.
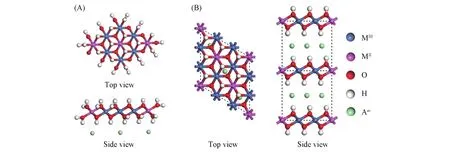
Fig.2 Cluster model(A)and periodicity model(B)of LDHs
在早期的计算模拟中,由于计算条件的限制多采用团簇模型进行计算[32,33].LDHs的团簇模型[图2(A)]通常用于对层板性质的分析,是从晶体结构出发,以最小的MO6结构基元组合为原子簇,以尽量少的MO6结构单元(不少于3个)共边组合来模拟LDHs层板结构.团簇模型构建的关键在于两方面:一是团簇整体的原子数目,需要尽可能反映出原晶体结构中原子的联接方式;二是团簇边界原子的处理方式,对于LDHs层板,通常采用端基加氢原子饱和的方式.团簇模型由于其原子数目少,计算上比较省时,在研究局域性质方面具有一定的优势;但是由于在某些方面不能完全反映晶体结构的整体性质,其应用受到一定的限制.
随着计算技术和计算机硬件能力的发展,近年来大多数关于LDHs的理论研究采用周期性固体模型[图2(B)][28,29,34],基于LDHs的晶体结构来构建,能够较为全面地反映晶体结构特征. 周期性固体模型将固体视为具有平移周期性的理想晶体,将固体中电子的运动视作电子在周期性势场中的运动,并引入能带理论对晶体的电子结构进行描述.但也因其模型大,涉及原子数目多,计算的周期会比较长.在实际计算时需要根据研究目的来确定用哪类模型最为经济和适用.
在模型的建立上,周期性模型根据LDHs实验上的结构参数.LDHs层板堆积方式有3R和2H两种,其中最常见的是3R方式.3R堆积方式的LDHs空间点群为r-3m,2H堆积方式的LDHs空间点群则为R-3H.晶胞参数为α=β=90°,γ=120°,而晶胞参数a,b和c则根据粉末X射线衍射(XRD)数据结合布拉格方程式(1)得到.值得注意的是,采用3R堆积结构的晶胞参数c是层间距的3倍,采用2H堆积结构的晶胞参数c是层间距的2倍[1].LDHs主要存在的特征衍射峰为(003),(006)和(110)等,通过(110)峰的位置,利用式(2)可以得到晶胞参数a(a=b);通过(003)峰的位置,利用式(3)可得到晶胞参数c.现将部分已报道的常见二元LDHs的晶胞参数列于表1,其它LDHs的晶胞参数见本文支持信息表S1.


Table 1 Experimental cell parameters of the reported binary MⅡ2M(Ⅲ/Ⅳ)⁃Cl⁃LDHs(polytype:3R)
1.2 计算参数
目前,对LDHs体系进行的研究主要基于密度泛函理论(DFT)和分子动力学(MD)方法.
1.2.1 DFT方法计算参数 当基于DFT对体系进行研究时,可调变的参数包括泛函、赝势、截断能、收敛限和k点等,且由于LDHs材料的结构特性,非键相互作用、自旋极化和Hubbard校正(DFT+U)等通常也需要被考虑.
在以LDHs材料为研究对象时,若采用周期性模型,可用Vienna Ab-initio Simulation Package,Material Studio(Castep module)等软件进行计算.通常参数可以设置为GGA/PBE泛函、Ultrasoft赝势、截断能340 eV以上.Yan等[57]研究了不同的交换关联泛函(LDA/CA-PZ,GGA/PW91,GGA/PBE和GGA/RPBE)、截断能(310,340和380 eV)搭配Ultrasoft赝势计算水镁石的晶胞参数,并与实验值相对比.综合发现,当截断能为380 eV时,LDA/CA-PZ计算的晶胞体积相对于实验值减缩了7.2%;而PBE和PW91高估了晶胞体积(4.5%~5.9%),且PBE相对于PW91在较低的截断能(340 eV)时,更贴近实验值;RPBE高估了晶胞参数,导致晶胞体积超过实验值19%.因此,GGA/PW91和GGA/PBE都是在LDHs计算中可以考虑使用的泛函,GGA/PBE在较低截断能下计算的晶胞参数与GGA/PW91相比更贴近实验值.截断能可根据具体的情况进行设置,必要时可以像上述工作一样进行测试,再做出符合情况的选择.
关于LDHs中非键相互作用的影响,Costa等[58](表2)分别采用PBE和revPBE-vdW(包含了范德华相互作用)2种泛函计算ZnAl-Cl-LDH的晶胞参数、金属键键长和金属氧键键长等;对比模拟值与实验值,发现revPBE-vdW过高估计了LDHs的晶胞参数,不适用于计算LDHs的结构.

Table 2 Calculated and experimental cell parameters of the optimized LDHs structure[58,59]
自旋极化(Spin polarized)和Hubbard校正(DFT+U)也是LDHs计算中需要考虑的问题,特别是涉及到含过渡金属的LDHs体系.开启Spin polarized可以对体系中的单电子数进行初始设置,有利于结构尽快地收敛.而Hubbard校正则是由于DFT采用了单电子近似,导致其对于强电子-电子相互作用的描述不准确而采用的一种校正方法[62].表3列出了常出现在LDHs中第一周期过渡金属的Ueff值.

Table 3 Ueffof the transition metal cations of LDHs
需要明确的是,Spin polarized和DFT+U并不是适用于所有的含过渡金属LDHs体系,需要根据计算的目的有选择性对这些参数进行设置.Moraes等[59](表2)采用3种交换-关联泛函GGA/PW91,GGA spin polarized以及GGA+U搭配Ultrasoft赝势计算了系列MgFe-LDHs的晶胞参数以及相关的电子性质.通过对比发现,3种泛函模拟的晶胞参数与实验值具有良好的一致性,其相对误差值均在可接受的范围内,GGA+U会高估晶胞参数a,b.因此在研究晶体结构时可采用最省时的GGA/PW91进行计算;对于差分电荷,3种泛函模拟结果相似;而涉及到电子态密度(DOS)的计算时,只有GGA+U能计算得出LDHs材料的半导体性质;计算总能量、振动结构及热力学性质时,GGA spin polarized则更为合适.
若采用团簇模型,可用Gaussian软件进行计算.Yan等[27]建立了LDHs八面体共边团簇模型[M2Al(OH2)9(OH)4]3+(M=Mg2+,Ca2+,Mn2+,Fe2+,Co2+,Ni2+,Cu2+,Zn2+,Cd2+),泛函设置为B3LYP,分别采用了6-31G(d)基组[M,Al,O,H采用全电子基组6-31G(d)]和混合基组[M采用LANL2DZ赝势基组,Al,O,H采用全电子基组6-31G(d)]对M=Mg2+,Ca2+的团簇进行几何优化,对比相关的参数发现两种基组的计算结果相似(表4).因此采用了更能节省计算资源的混合基组搭配B3LYP泛函进行后续的系列研究[69].
Hu等[70]为研究使用反应耦合分离技术从盐湖卤水中分离出Mg和Li的边界条件与初始Mg/Al比和Li/Al比之间的关系,建立了系列Mg/Al比为1~7和Li/Al比为1~7的LDHs团簇,采用B3LYP/LANL2DZ基组进行几何优化,采用B3LYP/6-31G(d)计算单点能.
随着计算技术的发展,出现了更多的新型泛函和基组,这将有利于LDHs团簇模型计算精度的进一步提升.依据不同的计算目标和计算能力,需要进一步平衡计算精度与计算用时之间的关系.

Table 4 Optimized Geometries of[M2Al(OH2)9(OH)4]3+[27]
1.2.2 MD方法计算参数 当使用MD方法对LDHs体系进行研究时,需要考虑的参数主要为力场、系综、温度与压力等.通常采用周期性模型,可用Material Studio(Forcite module),GROMACS等软件进行计算.
力场的选择对模拟结果的准确性起关键性作用.目前在LDHs模拟中使用较多的CLAYFF力场是Cygan等[71]针对黏土材料研究出来的一种力场,其中大多数参数是根据阳离子黏土材料得出的,并不完全适合LDHs类化合物的分子模拟.CVFF,Dreiding等力场也被应用于LDHs的模拟[72~74],但由于缺少金属离子在八面体场中的力场参数,计算时均需对其中的某些数据进行改写.鉴于此,Zhang等[75]采用“双阱势”模拟LDHs主体层板中的六配位金属结构,得到了多种金属离子的力场参数,建立了适用于LDHs层状材料的通用分子力场LDHFF.LDHFF力场的势函数表达式为
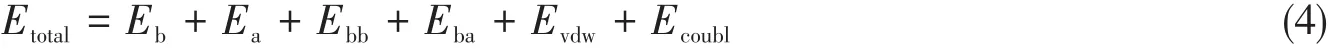
式中,Eb为键伸缩项;Ea为成键作用项;Ebb和Eba为交叉作用项;Evdw和Ecoubl为非键作用项.其中,
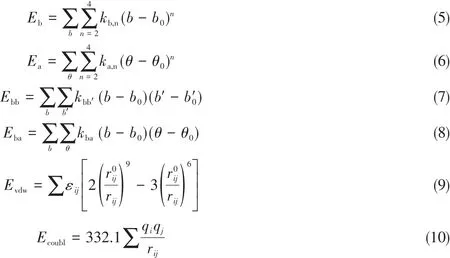
式中,kb,n(kcal·mol—1·Å—4)为键伸缩相互作用的振动常数(1 kcal=4.19 kJ,1 Å=0.1 nm);b0(Å)为平衡键长;ka,n(kcal·mol—1·rad—2)为键角弯曲的常数;θ0(°)为平衡键角;kbb′(kcal·mol—1·Å—2)和kba(kcal·mol—1·Å—2·rad—2)为交叉项中的耦合常数;εi(kcal/mol)为弱相互作用的范德华势阱深度;(Å)为作用半径;qi(e)为力场电荷.
LDHFF力场有很好的通用性,基本包含了所有LDHs体系主体层板中的金属离子;在分子动力学模拟中,层间阴离子的总电荷为化学式电荷(Formula charge),其中单个原子的电荷是通过DFT计算的Mulliken布居数分析电荷值.层间客体阴离子元素的力场参数是来自PCFF力场.通过Ewald方法计算带电原子间的库仑力作用[75];通过Spline cutoff方法计算原子/分子间范德华力.
LDHFF可以维持长时间动力学模拟过程中主体层板的结构稳定性,显著拓展了LDHs分子动力学模拟的时空尺度,目前已被多次应用于LDHs材料层间客体取向结构和分布的模拟和预测[76~79].运用LDHFF力场,Pan等[76]模拟了H-LDH/CTS主客体的相互作用以及氧气在H-LDH/CTS中的热运动,证实了H-LDH在提高氧气阻隔性能方面起着关键作用;Dou等[77]证实了CA/LDH中CA与LDH间的氢键网络是抑制氧气扩散的因素,并在此基础上,模拟了CO2吸附对LDH/PPA的氧气阻隔性质的影响[61],证实了吸附的CO2导致了薄膜的超低透氧性.这些结果为开发新型气体阻隔材料提供了理论指导.Xu等[80]模拟了荧光分子与LDHs的相互作用,为基于LDHs插层方法解决荧光分子团聚的问题提供了很好的借鉴.这些实践应用表明,LDHFF力场为新型LDHs功能材料的构筑和设计提供了理论指导和模拟依据,是对LDHs材料分子模拟理论的重要发展.
一个系统的状态变量有能量(E)、焓(H)、粒子数(N)、压力(P)、应力(S)、温度(T)和体积(V),根据固定状态变量的不同,生成的不同统计集合即为系综.MD模拟总是在一定的系综下进行.平衡系综一共有4种,分别是微正则系综(NVE)、正则系综(NVT)、等温等压系综(NPT)和等压等焓系综(NPH),分别适用于不同体系.一般气体的扩散过程中,系统的温度与压力为定值,通常采用NPT系综进行研究,因此在研究LDHs/聚合物复合材料的气体阻隔能力的相关工作中,使用的系综为NPT[76,77].而对于LDHs的插层与分离的相关工作,由于体系的粒子数目、体积与温度不变,通常采用NVT系综进行模拟.Lv等[81]利用MD方法,研究将光敏染料甲基橙插入Mg2Al-LDHs采用的系综是NVT.Wang等[82]利用MD来解释LDH薄膜中氨基酸的插层和分离机制也采用了NVT系综.
2 主体结构与特性
LDHs的晶体结构特征主要由主体层板的元素性质、客体阴离子的种类和数量、层间水的数量及层板的堆积形式所决定.在LDHs的结构中,金属离子组成、排布及层板堆积方式直接关系到层板电荷的分布、电子性质以及拓扑转变和记忆效应等,进而影响到层间客体阴离子的成键、反应活性、位置、取向和层板表面性质(如催化活性)等.
对LDHs的主体结构与特性进行研究主要是基于LDHs的团簇和周期性模型,利用DFT方法对结构的电子性质等进行计算;对于拓扑转变和记忆效应则是采用了基于DFT的MD方法对周期性模型中原子的迁移进行模拟.
2.1 主体层板的元素类型
关于LDHs的主体结构构筑,长期以来一直沿用与镁离子半径相近的离子可引入层板的经验判据[83],但其对诸多实验现象无法给出合理解释.如,Ca2+和Cd2+与Mg2+半径相差很大(0.1和0.095 nm),但它们可以形成稳定的CaAl-LDHs[84,37]和CdAl-LDHs[85]主体层板;而 Pd2+和 Pt2+与Mg2+半径相近(0.086和0.080 nm),却难以形成PdAl-LDHs或PtAl-LDHs[86,87]的主体层板.
针对上述问题,Yan等[88]根据LDHs主体层板是由二价和三价金属离子的八面体共边形成的特点,提出由最小结构基元MO6八面体的结构稳定性来判断LDHs主体层板稳定性的设想,采用DFT的理论方法,对合成LDHs可能涉及的二价和三价金属离子形成的MO6八面体基元进行了计算(图3).基于[M(OH2)6]n+(M为金属离子,n=2,3)团簇模型,将∠O-M-O偏离90°的大小定义为八面体变形度表示大于90°键角的平均值;表示小于90°键角的平均值]. 可根据八面体变形度的大小将计算涉及的金属离子分为3类:第一类八面体为正则结构(θ=0°~1°);第二类八面体变形较小(θ=1°~10°);第三类八面体变形较大(θ>10°).
对MO6八面体的结构性质如M—O键长、O—M—O键角及其变形度、自然键轨道(NBO)以及结合能等性质进行详细分析,并与实验结果对比表明,金属离子是否能稳定地引入层板与其所处的分类有关,总体来说,引入的稳定性次序为第一类>第二类>第三类.
第一类金属离子引入LDHs材料中可以得到稳定的LDHs层板结构,且二价和三价离子之间易于相互组合形成二元LDHs层板;第二类金属离子倾向于作为多元LDHs中的其中一个组分;而第三类金属离子由于形变较大,则难以引入LDHs中形成稳定结构.此研究提供了一个全新的判定方法,即当引入层板的金属离子与Mg2+有相似的离子半径时,其变形度相对于离子半径对结构的影响更大;而对于引入层板的金属离子半径远大于Mg2+时,则需要考虑其优先的配位倾向[88].以此八面体变形度为依据,可以对与镁离子半径相近经验判据无法解释的实验现象进行很好的解释:Ca2+,Cd2+与Al3+同属于第一类,而Pd2+和Pt2+则属于第三类,故前者可以形成LDHs结构,而后者不能形成.
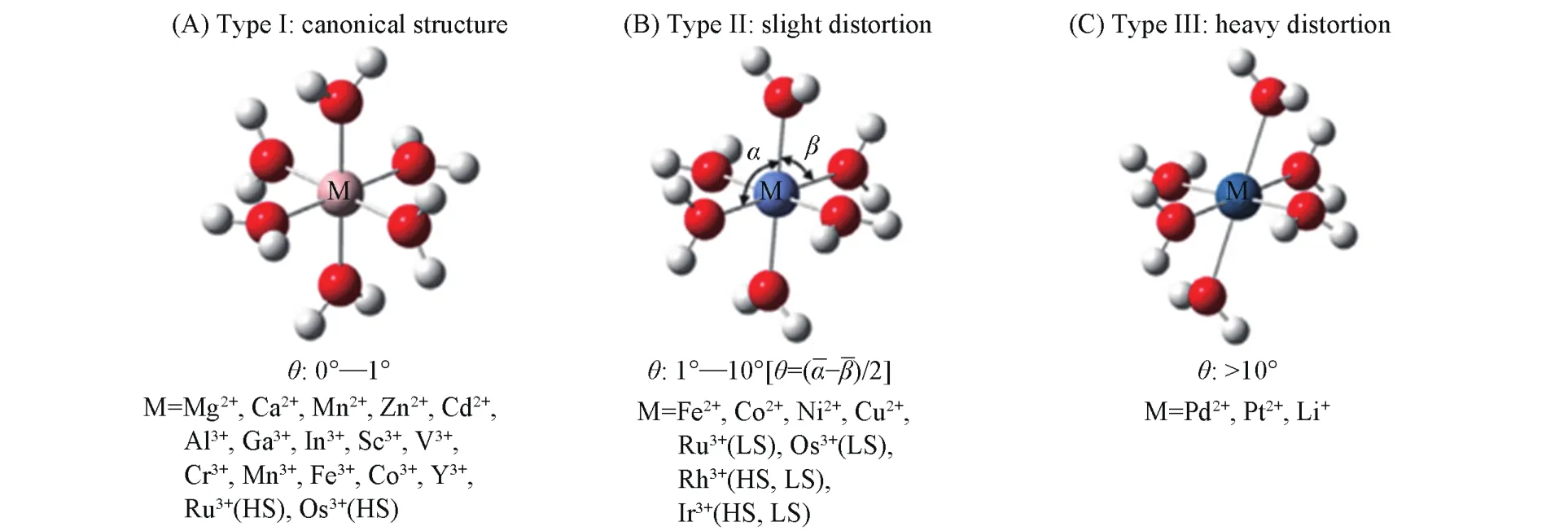
Fig.3 Optimized structure of three types of[M(OH2)6]n+clusters[88]
八面体变形度判据反映了金属离子的配位环境,这一因素较金属离子半径对其是否能形成六配位的八面体结构更具有决定性意义,为合成新型的LDHs提供了理论指导.
Yan等[27]进一步建立了包含LDHs基本结构信息的最小团簇模型[MII2Al(OH2)9(OH)4]3+(图4),系统研究了团簇的几何参数(键长、键角)、自然键轨道(NBO)、三中心桥位羟基的伸缩振动频率[ν(O3—H)]以及金属离子的价电子排布、结合能、姜-泰勒效应等直接影响LDHs的结构性质与相对稳定性的参数,进一步说明不同的MⅡ的电子结构对团簇性质的影响.结果表明,计算得到的团簇模型的几何结构以及ν(O3—H)与模型中MII的电子结构,如价电子构型、自然键轨道、自然电荷传输以及键级相关.计算所得的结合能与实验所得相应LDHs的相对稳定性也具有一致性.故相对于离子半径大小,金属离子的电子结构对LDHs材料的结构性质与相对稳定性的影响更大[27].

Fig.4 [MII2Al(OH2)9(OH)4]3+cluster model(A),the linkage around the three⁃centered bridging OH group(B),the relationship between zero⁃point corrected binding energy(ZPE)ΔEb(1 kcal=4.19 kJ)of the clusters and the atomic number of MII(C)[27]
在此基础上,Yan等[89]将层板中引入阳离子的可能性和该元素在周期表中的位置进行关联,指出在层板中的阳离子的电子构型和配位性质会引起LDHs的基本性质,如局域配位构型、稳定性、电负性、能带结构的周期性变化.这些周期性变化将直接影响LDH在固体碱催化和光催化等方面的应用.
2.2 主体层板元素的组成比例
MII/MIII(R)比在LDHs结构的形成中具有重要的作用.LDHs层板的电荷密度是由R值决定的,因此通过改变R值可以对层间阴离子的数目和排列方式进行调控,从而实现LDHs的可控制备.Yan等[69]基于DFT方法对不同R值(R=2~6)的ZnAl-LDHs团簇进行研究,通过系统计算团簇模型的几何参数以及形成能,分析不同R值下的团簇构型以及Al3+和Zn2+阳离子周围的成键,发现当R<5时,在相应的ZnRAl-LDH团簇中,Al3+相对于Zn2+在决定微观结构性质、构成以及键合稳定性中发挥了更重要的作用;当R≥5时,Zn2+成为了主导因素.不同的R值对系统稳定性的影响也很大(图5),LDHs相在R=2~5时可以保持稳定的状态,而且在此范围内R越小越稳定;而当R≥5时,LDHs相存在的稳定性下降.
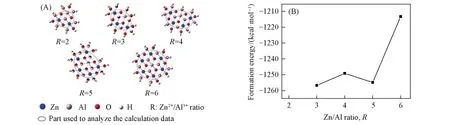
Fig.5 ZnAl⁃LDHs clusters with R(Zn2+/Al3+)in the range of 2—6(A)and relationship between the formation energy Ef(1 kcal=4.19 kJ)of the ZnRAl⁃LDHs(R=3—6)clusters and R(B)[69]
Yan等[57]基于DFT以及虚拟晶体近似(VCA)使用平面波赝势,对金属离子比R=2~5的MgAl-Cl-LDHs周期性模型的几何构型、晶格能量以及态密度进行了研究.对于相同大小的MgAl-Cl-LDHs晶胞,R变大会不可避免地导致更多的Al(Ⅲ)—O—Al(Ⅲ)键的产生.从几何构型角度,Al(Ⅲ)—O—Al(Ⅲ)键数目和主层板电荷密度的增多,导致了金属阳离子之间更强的静电排斥力、更长的Al—O键键长、更小的O—Al(Ⅲ)—O键键角以及层间距;对于晶体能量,模型中存在的Al(Ⅲ)—O—Al(Ⅲ)键导致了局域电荷密度上升,从而使得晶体整体能量的上升,导致结构不稳定(图6).综上所述,当R值在一个较小的范围内时,为获得稳定的LDHs结构,应尽量避免M(Ⅲ)—O—M(Ⅲ)键存在.同时,3种堆积方式下,无序性Mg-Al-Cl-LDHs的稳定性均随Mg/Al比的增大而增大.当R值相同时,3种堆积方式下的晶格能大小顺序为3R>2H>1H.说明3R堆积方式最稳定,这一结果同样符合实验事实.
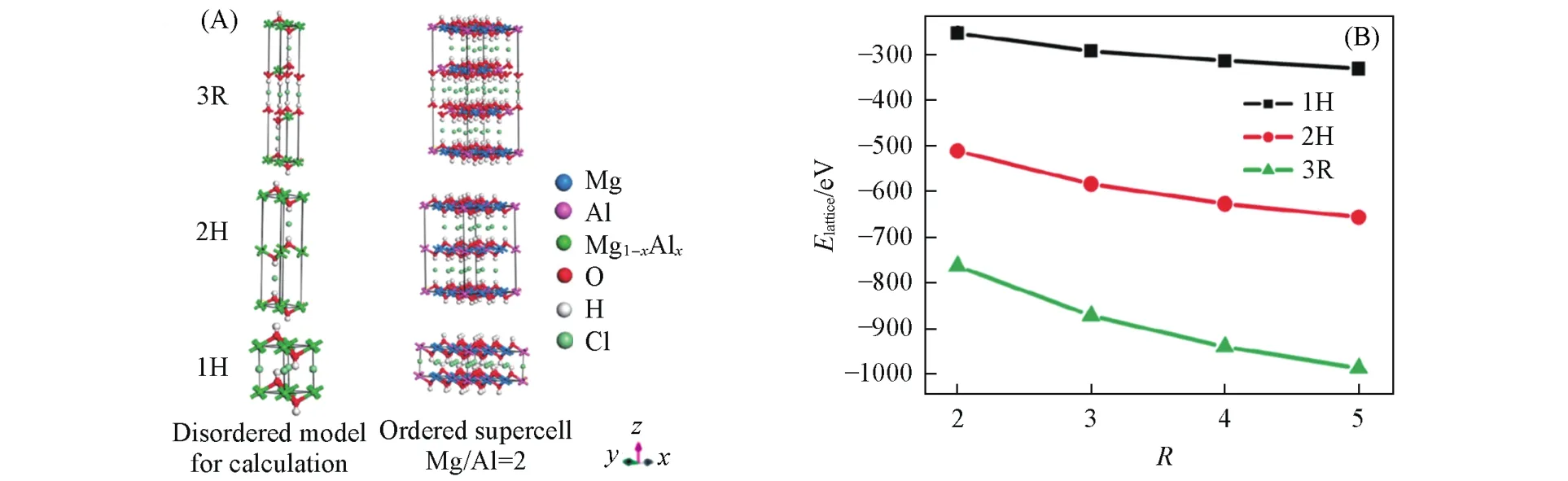
Fig.6 Periodic disordered computational model formulated as Mg1-xAlx(OH)2Clxand ordered 3×3×1 supercells(Mg/Al ratio=2)for MgAl⁃Cl⁃LDHs with different stacking sequences(3R,2H and 1H)(A)and lattice energy of the disordered MgAl⁃Cl⁃LDHs model stacking in different sequences as a function of R(B)[57]
R(MII/MIII)的改变也会导致LDHs电子性质的改变.Wang等[90]使用密度泛函理论研究了R=2~4的ZnTi-LDHs,发现随着Ti含量的增加,导致LDHs带隙减小,紫外吸收能力增强.Ding等[91]使用DFT+U方法以及MD方法研究了系列Ni3—xCoxAl-LDHs(x=0,1,2,3)的离子导电与去质子化能力,发现Co,Ni阳离子的共存使LDHs具有更大的层间距,激发了OH—的扩散,且NiCo2Al-LDHs具有最高的比电容.
2.3 电荷分布特征
LDHs层板电荷的分布对于进一步开发LDHs的催化、电学方面的功能性具有重要意义.Liu等[34]根据前期研究得到,(003)晶面是LDHs优先暴露晶面,并对(003)晶面的电荷分布进行了研究,基于DFT方法计算了系列MgAl-LDHs的电荷差分性质(图7).差分电荷代表的是不同粒子间的电荷再分配.图7显示的是R=2时,Mg2Al-A-LDHs体系层板的差分电荷图.其中红色代表电荷积累,蓝色代表电荷消耗.可见,不同阴离子插层的LDHs层板的差分电荷图相近,表明金属阳离子的电荷转移是相近的,说明插层阴离子对LDHs层板的电荷转移几乎没有影响.Mg2+周围出现较少的电荷消耗(浅蓝色),Al3+周围则出现较多的电荷消耗(蓝色),这表明在形成LDHs金属层板时,相对于Mg2+,Al3+的电子密度减小较明显,这与Al3+本身拥有更高的正电荷是一致的.Mg2+和Al3+周围区域的差分电荷图没有重叠,表明Mg—O和Al—O键为离子键.

Fig.7 Electron density difference of layer in Mg2Al⁃A⁃LDHs[34]
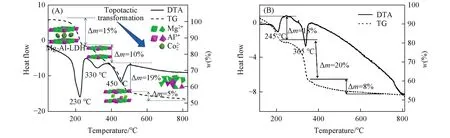
Fig.8 Simulation of the thermal decomposition process of the MgAl⁃LDHs(A)[94]and NiAl⁃LDHs(B)[95]
2.4 拓扑转变与记忆效应
LDHs材料在一定温度下可以发生拓扑转变,即反应物和生成物存在着某种不变性;且部分LDHs具有记忆效应,即由焙烧产生的复合金属氧化物(Mixed Metal Oxides,MMO)在室温的条件下与溶液相混合时可恢复LDHs层状结构.对LDHs的结构拓扑转变和记忆效应的机理进行研究对制备高度分散的高效纳米催化剂具有重要意义,但实验方法很难从原子的水平上理解LDHs发生拓扑转变和记忆效应的机理.根据前期实验研究,大多数的LDHs经煅烧导致的拓扑转变过程有3个步骤:(1)LDHs层板吸附水分子以及层间存在的水分子的蒸发;(2)层间的阴离子的热分解;(3)层板上羟基的脱出[92,93].Zhang等[94]和Meng等[95,96]采用基于DFT的第一性原理分子动力学模拟(AIMD)方法并结合热重(TGDTA)分析研究了MgAl-LDHs,ZnAl-LDHs,NiAl-LDHs和MgFe-LDHs分别在其关键性温度热分解(MgAl-LDH:330,450及800℃;ZnAl-LDH:273,800℃;NiAl-LDHs:365,800℃;MgFe-LDHs:380,800℃)升温过程的拓扑转变机理及记忆效应(图8).模拟结果表明4种LDHs均通过单齿配体进行分解.MgAl-LDHs在330℃下发生层间阴离子的分解,并释放出气态水分子和CO2,450℃下发生主体层板脱水的第一阶段;而MgFe-LDHs,ZnAl-LDHs和NiAl-LDHs层间碳酸根离子的分解和部分层板脱羟基分别发生在同一温度下.
将主体层板逐渐脱水过程中的LDHs模型用CLDHs表示,并用数字代表脱除的水分子个数,如CLDHs-1代表脱除1个水分子的产物,CLDHs-6代表脱除6个水分子的产物(图9).通过向拓扑转变过程中的6个脱水模型(CLDH-1~CLDH-6)中插入CO2和H2O,测试不同模型的记忆效应,发现CLDH-1~CLDH-5模型均可以重新构成与LDHs结构相同的八面体层,CLDH-6中没有观察到结构重构过程.将LDHs焙烧分解过程中结构变化层板的金属离子的分散度(σ)定义为拓扑不变量,结合均方根位移(RMSD),反映金属离子在LDHs(001)面和(120)面的迁移规律[94],定义式如下:
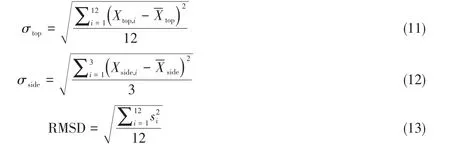
式中,σtop表示LDH(001)晶面中金属离子的分散度,将LDH(001)晶面均分为12个六边形区域(图9),并保证理想情况下金属离子占据六边形的中心;Xtop,i表示金属离子分布在第i(i=1,2,3,4,…,12)个六边形区域内的总个数;表示平均每个六边形区域内金属离子分布的数量[对于LDH(001)面,=1];σside表示LDH(120)晶面中金属离子的分散度,沿c轴方向将LDH(120)均分为3个区域(图9);Xside,i表示金属离子分布在第i(1,2,3)个区域内的总个数;表示平均每个区域内2价或3价金属离子的数量;si(Å)表示第i(i=1,2,3,…,12)个金属离子与该金属离子初始位置之间的空间位移值,通过RMSD(Å)可观察金属离子偏离其初始位置的程度.
通过计算金属离子的分散度和均方根位移,发现金属阳离子Mg2+,Al3+在(001)晶面内几乎没有发生移动,只在c轴方向上移动[94].
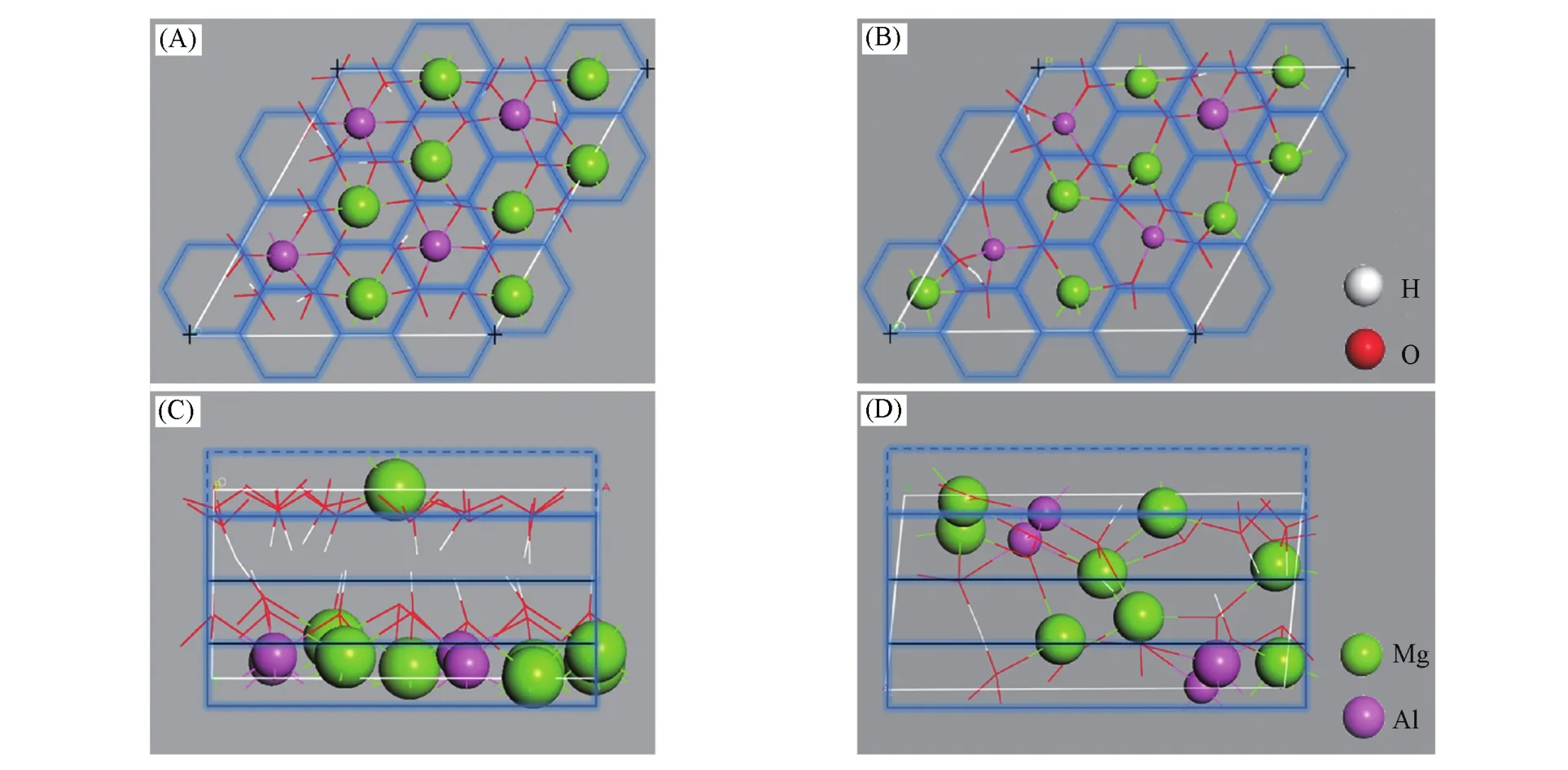
Fig.9 Top⁃view of CLDH⁃1(A)and CLDH⁃6(B);side⁃view of CLDH⁃1(C)and CLDH⁃6(D)[94]
研究发现,在层板脱羟基的过程4种LDHs并不相同.MgAl-LDHs,NiAl-LDHs,MgFe-LDHs中金属离子(Ni2+,Al3+,Mg2+,Fe3+)在层板[LDHs(001)晶面]方向上其拓扑不变量(层板方向迁移度)几乎保持不变,而在LDHs(120)晶面上金属离子的迁移较大.而ZnAl-LDHs中的Zn2+,Al3+的拓扑不变量无论是在LDHs(001)晶面还是在LDHs(120)晶面上都发生了明显的迁移.进行LDHs记忆效应过程的动力学模拟,发现在室温的条件下,MgAl-LDHs,NiAl-LDHs,MgFe-LDHs能恢复层状结构,而ZnAl-LDHs不能恢复,因此MgAl-LDHs,NiAl-LDHs,MgFe-LDHs在此温度下有记忆效应,而ZnAl-LDHs则没有记忆效应.说明记忆效应与拓扑不变量密切相关.同时,ZnAl-LDHs结构中金属离子完全变为四配位,很难恢复到六配位,而NiAl-LDHs,MgFe-LDHs在M-CLDH-1结构中只是部分金属离子变为四配位或者五配位,因此金属离子能较容易地恢复到六配位.在800℃下发现所有LDHs结构已经完全坍塌,形成多孔结构.模拟结果与实验XRD数据高度一致.此研究从原子水平上理解了不同LDHs整体结构从热分解的起始阶段到完全脱水的演变过程,对于认识LDHs热致拓扑转变机理及记忆效应,及设计高分散催化剂提供了有益的理论信息和指导[95,96].
3 客体结构特性
为获取不同功能性的LDHs材料,实验中常采用阴离子交换法将具有不同功能性的阴离子插层进入LDHs[97].因此,客体阴离子在基于LDHs的功能性材料中起着关键作用.但是,由于LDHs低结晶度以及其固有的结构紊乱,通过现有的实验手段很难确定层间客体的取向以及相关的性质.借助计算手段,人们已对于层间客体的溶胀性质、阴离子排布方式、离子交换性质、客体相互作用等进行了研究[60,98,99].
对客体结构特性的研究常基于周期性模型,采用DFT方法以及MD方法进行研究.

Fig.10 Possible packing styles of interlayer water molecules of α⁃Ni(OH)2and Ni/Al⁃LDHs[100]
3.1 层间客体的取向和分布
通过理论计算可以对LDHs层间阴离子的取向、在层间的排布、层间阴离子的电子结构等进行研究.Li等[100]构建了包含水分子和不同层间客体的阴离子(F—,Cl—,Br—,OH—,N,C和S)的NiAl-LDHs,并采用修饰过的cff91力场进行MD模拟.通过模拟将含有不同客体NiAl-LDHs的层间水分子的堆积模式分为3种类型(图10):类型1,当层间阴离子为N,C,Cl—,Br—和OH—时,插层水分子和阴离子紧密堆积并平行于层板;类型2,当层间阴离子为S时,层间水分子分布比较松散;类型3,当层间含水量较多时,水分子在层间呈双层排布.2007年,Kumar等[101]运用MD方法探究了不同种类有机羧酸根插层的MgAl-LDHs的结构和能量性质,结果显示,所有的一元羧酸根都是平行于层板分布于层间,甲酸根中的—COO—平行于层板排布,丙酸根中的—COO—垂直于层板,而乙酸根中的—COO—的取向是无序的.
Liu等[34]基于量子力学方法,采用DFT方法模拟了不同阴离子插层的MgAl-LDHs,发现当阴离子价态相同时,阴离子在层间的排布方式与其半径大小密切相关,半径越大,越倾向于倾斜或垂直于层板排布以及阴离子的尺寸越大,电荷越低,层板电荷对其取向的影响越大.
3.2 客体间的相互作用
层间阴离子的功能性影响LDHs的功能性,另外,LDHs的晶体结构中通常会含有水分子,虽然这些插层水分子可以通过热处理去除,然而,由于其本身具有高度的吸湿性,LDHs可以从大气或溶液中吸收水分子重新水合[102].水分子可以和层板的羟基形成氢键来增强LDHs的稳定性,同时也可以和阴离子相互作用[103~105].LDHs类化合物的某些性质,如层间距、阴离子交换能力和催化活性,均会受到其结构中水分子数量和阴离子种类的影响[106].Xu等[80]基于LDHFF力场,利用MD模拟方法对染料和不同链长烷基磺酸盐(CnH2n+1SO3,n=5,6,7,10,12)表面活性剂分子共插层ZnAl-LDHs结构进行了模拟,发现共插层分子尺寸相近时,客体与客体之间相互作用最大,LDHs的结构最为稳定,并提出了2个抑制染料在LDHs中聚集的方法:选择一种与染料大小相近的表面活性剂作为共插层,或者增加LDHs基质中三价阳离子的含量.
Liu等[34]利用DFT方法分析了不同阴离子插层的Mg2Al-LDHs层间物种的差分电荷(图11),发现层间阴离子与水分子之间的相互作用主要来源于静电力(根据氢键定义:层间物种的X…H或O…H的氢键作用也属于静电相互作用[107]).
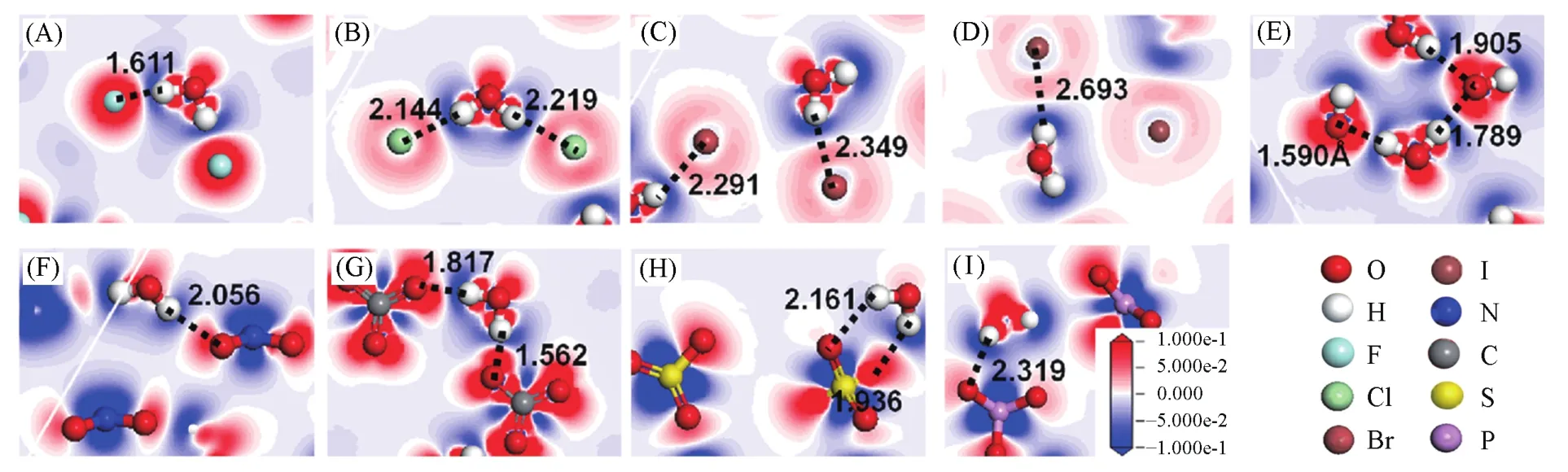
Fig.11 Electronic density difference of interlayer species of Mg2Al⁃A⁃LDHs interlayered with different anions(A=F-,Cl-,Br-,I-,OH-,N,C,Sand P)under the top view,along with the distances between the H atom(H)of water and halogen anions(X)(dX···H)or O atoms within oxygen acid anions(dO···H)(in Å,1 Å=0.1 nm)[34]
3.3 层间距的影响因素
实验中通常可以通过XRD得到LDHs的层间距.影响层间距的因素有很多,如反应原料的配比、合成的条件、层间阴离子的种类和排布方式、水分子的数量等.Leitão等[58]基于DFT方法模拟了不同阴离子插层的ZnAl-LDHs的结构和性质,通过结构分析可以得到F-,OH-,Cl-和Br-插层的ZnAl-LDHs晶胞参数c分别为22.24,22.70,23.47和23.76 Å(1 Å=0.1 nm),层间距大小与层间阴离子的大小相关.Liu等[34]运用DFT方法模拟了不同层板比例对不同阴离子插层的LDHs层间距的影响(图12),揭示了不同层板比例会影响层间距.且一般情况下,电荷相同的阴离子半径越大,尺寸相近的阴离子所带电荷越小,层间距越大.以及层间阴离子的排布方式也很大程度上影响层间距,并模拟了层间不同的含水量对层间距的影响,揭示了层间较多的水分子使得层间距增加.Ni等[108]采用MD方法模拟了谷氨酸插层的ZnAl-LDHs(Glu-ZnAl-LDHs)的超分子结构,研究了水分子的数量对层间距的影响,研究发现,当水分子数量小于8时,层间距缓慢增加,当水分子数量大于等于8时,层间距与水分子数量呈线性关系.
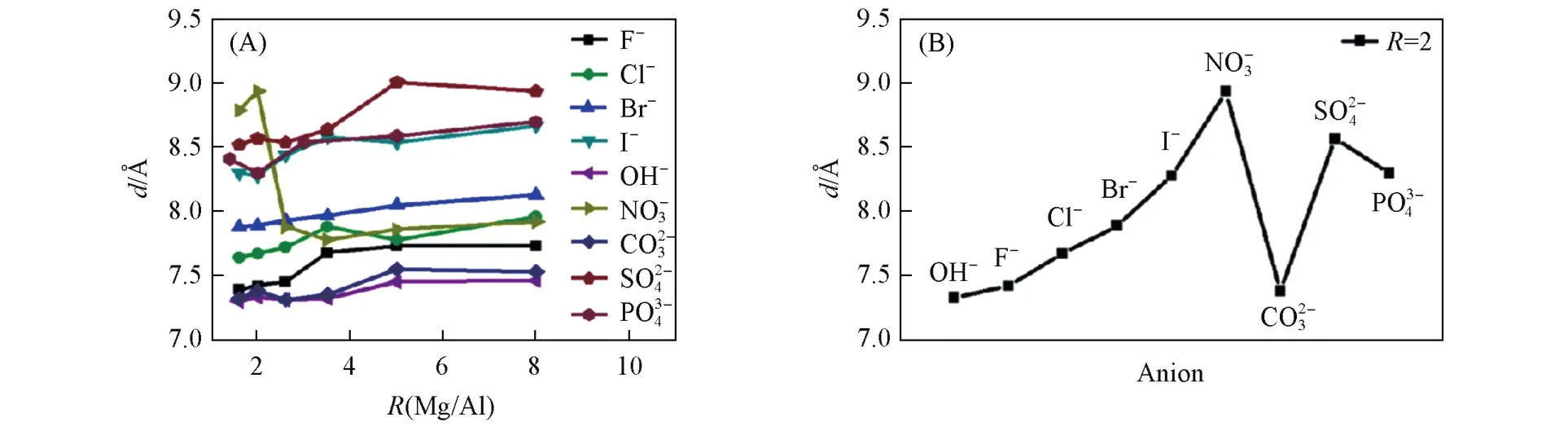
Fig.12 Interlayer distance(d)of MgAl⁃A⁃LDHs with different anions and different R(Mg2+/Al3+)(A)and Mg2Al⁃A⁃LDHs with different anions(B)[34]
3.4 离子交换性质
LDHs的层间阴离子与层板间存在静电作用,这种弱相互作用力使得层间阴离子具有可交换性,可将目标阴离子通过离子交换方式引入层间得到所需的功能性LDHs材料[109].与阴离子交换树脂相比,LDHs具有较大的离子交换容量,且具有耐高温、耐辐射、体积小、密度大等优点,目前已经在废水处理、荧光、生物医药等方面有了一定应用[110~112].阴离子与LDHs的结合能力可用于理解不同阴离子交换能力的强弱.Li等[101]运用MD方法证实了NiAl-LDHs中不同阴离子的离子交换作用.Liu等[34]采用DFT方法探究了不同层板比例的LDHs与不同层间阴离子的结合能力,通过计算结合能可以得到阴离子所带电荷越高,越易插层;电荷越低,越易被交换(三价>二价>一价),且阴离子所带电荷相同时,尺寸越小,越易插层.因此,就离子交换能力而言,阴离子半径比较大的容易被交换出来,这种类型的含低价态客体的LDHs可以作为交换前驱体.
4 插层结构系统特性
对LDHs插层结构的研究,主要是基于周期性模型,对其采用DFT方法或MD方法来进行探究.
4.1 主客体相互作用
LDHs的主体层板带有一定数量的正电荷.因此,需要在层间引入阴离子来平衡电荷以确保LDHs材料保持电中性.LDHs主客体之间存在着多种弱相互作用,包括静电力、范德华力以及氢键.主客体之间的相互作用会影响LDHs中阴离子的排布方式、层间距等结构性质、离子交换反应的性能、热稳定性能等.Leitão等[112]基于密度泛函理论和周期性边界条件对LDHs化合物进行从头计算,结果表明,层间阴离子为C的层间距比Cl-的层间距小,可归因于C与层板的相互作用力更强,并从差分电荷和振动分析进一步验证了此结果.Moraes等[59]基于DFT方法,绘制了含有不同层间阴离子(N,C,HP)的MgFe-LDHs的差分电荷图(图13),研究了层板、层间阴离子、层间结合水之间的相互作用.结果发现,层间阴离子C和HP与层板之间有更强的电荷转移.且在[Mg-Fe-HPO4,Na]中,HPO不仅与层板之间有强烈的相互作用,而且与水合Na+之间也存在强相互作用,说明了与HP进行阴离子交换比与N进行交换更加困难.该课题组[113]运用DFT方法模拟研究了水合前后十二烷基硫酸酯(DDS)插层的ZnAl-LDHs结构和性质变化.电子分析结果表明,靠近阴离子头的水分子倾向于将电荷转移分布在LDHs层板上,可以确定水分子与层板之间存在相互作用,而去水合后虽然主客体存在相互作用,但DDS发生结构弯曲促使层间距减小.Yan等[114]对具有对称性的邻菲啰啉钌配合物[Ru(dpds)3]4—在LDHs层间的行为进行了DFT计算研究,结果表明,[Ru(dpds)3]4—与LDHs带正电荷层板相互作用导致阴离子的对称性大大降低,S-Ru-S角度由原来的71.0°增加到110.4°,并解释主客体的相互作用是偏振光致发光的原因.Yan等[34]基于DFT方法分析了不同阴离子插层的Mg2Al-LDHs的主客体相互作用,通过差分电荷分析层板与层间阴离子的主客体相互作用主要来自于主客体间静电力,包括X…H,O…H之间的氢键作用,且高价阴离子与层板的相互作用比低价阴离子更强.
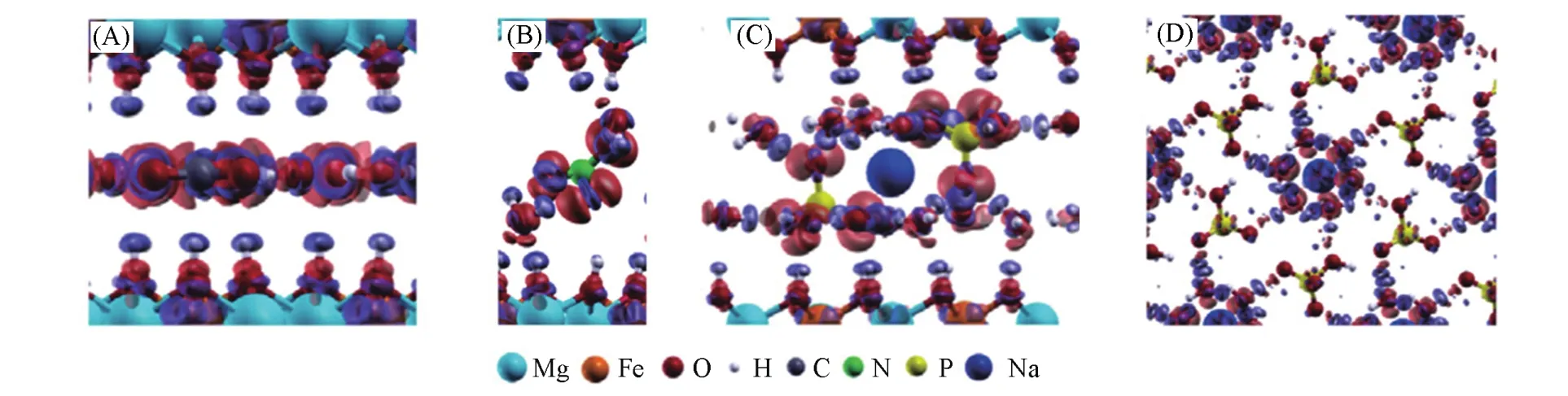
Fig.13 Electronic density difference of[Mg⁃Fe⁃CO3](A),[Mg⁃Fe⁃NO3](B),[Mg⁃Fe⁃HPO4,Na](C),and hexahydrated sodium(D)in the interlayer region of[Mg⁃Fe⁃HPO4,Na]with the HPanion,using GGA calculation[59]
The blue region presents the depletion of electronic density,and the red region indicates the increase of electronic density.Copyright 2016,American Chemical Society.

Fig.14 Band edge placements of MIIMII⁃LDHs(MII=Mg,Co,Ni,Zn;MIII=Al and Ga)[28]
4.2 能带结构与光催化性能
为深入研究LDHs在光功能材料、电学材料和催化剂方面的应用,对其能带结构的分析和研究具有重要的指导意义.能带理论认为晶体中多个电子的共同作用使单能级分裂成N个看似连续的能级,而这些能级构成了能带.因此能带结构反映了晶体的诸多信息,如导电性、带隙、能带边缘位置等,对其进行计算有利于明确材料的构效关系.Xu等[28]使用Hubbard方法校正的密度泛函理论计算了MIIMIII-和 MIII/IV为过渡金属的MIII=Cr,Fe;MIV=Ti;n=2,3,4;A=Cl-,NO3-,CO32-)的电子结构,得到了本征电子性质:能带结构、电子态密度、表面功函数、导带和价带边缘相对于真空能级的位置(图14),以此对其反应可行性以及反应路径进行预测.如,根据LDHs导带和价带相对于真空能级的位置,可以得到CoAl-LDHs,NiAl-LDHs,ZnAl-LDHs,NiGa-LDHs催化氧气生成反应(OER)的驱动力;而MgAl-LDHs,MgGa-LDHs和ZnGa-LDHs不具备氧气生成反应的驱动力.基于此计算了CoAl-LDHs,NiAl-LDHs,ZnAl-LDHs和NiGa-LDHs催化氧气生成反应的四步基元反应,并得到了每一步基元反应的Gibbs自由能变,确定了氧气生成反应的速率控制步骤.通过计算得到CoAl-LDHs,NiAl-LDHs,ZnAl-LDHs和NiGa-LDHs氧气生成反应的吉布斯自由能垒分别为0.65,0.60,1.10和0.94 eV,并将其与驱动力比较,发现只有CoAl-LDHs能够在光照下通过驱动力克服反应的吉布斯自由能垒自发生成氧气.而计算MIII/IV为过渡金属的MIII/IV-LDHs发现,Co2Fe-Cl-LDHs,Ni2Ti-Cl-LDHs,Zn2Ti-Cl-LDHs,Zn2Cr-Cl-LDHs及NinCr-A-LDHs能够在光照下通过驱动力克服反应的活化能自发析氧,此计算结果与实验观测一致[28,29].此研究提出了通过计算手段筛选LDHs作为光催化OER反应催化剂的方法.
除光催化OER反应,LDHs也可应用于光催化分解反应[115].Xia等[116]探究了具有不同层间阴离子的 ZnCr-M-LDHs(M=F—,Cl—,Br—,I—,NO3—)分解六氯苯的能力.通过DFT计算了不同LDHs的能带结构、电子态密度、氢键等,推断出其结构稳定性顺序为LDHs-NO3>LDHs-F>LDHs-Cl>LDHs-Br>LDHs-I,而光降解性能的顺序则相反.
LDHs材料因其结构简单、能带可调的特点,通过能带调控将其和其它氧化物、硫化物等相结合形成异质结,用于抑制光催化反应中光生电子和空穴快速复合,从而提高催化剂的光催化性能.Xia等[117]通过计算分析核-壳结构CeO2@CoAl-LDHs的异质结结构,对催化甲醛气降解的光催化机理进行推断.发现异质结中形成了电场强度方向为从CoAl-LDHs到CeO2的内置电场,在其促进下可以有效地抑制光生电子与空穴的复合.Wu等[118]通过剥离-再堆积技术制备CdS/ZnCr-LDHs的异质结,形成了层层自组装结构,对其进行了能带分析.形成的异质结结构具有可见光响应.光生电子从更加负的CdS导带传输到邻近的ZnCr-LDHs的导带.与此同时,光生空穴从ZnCr-LDHs的价带(2.39 eV)传输到CdS的价带(1.97 eV).在CdS和ZnCr-LDHs中存在的协同效应促进了光生载流子的传输与分离.
Li等[119]针对不同主体阳离子导致的LDHs电化学行为的协同效应进行了研究,发现与单过渡金属相比,双过渡金属的LDHs具有更低的带隙能和更高的电导率.带隙能量的降低和电导率的提高应归因于不同过渡金属原子的3d轨道间的杂化.Lv等[120]使用DFT方法对含有不同M2+的CuMAl-LDHs的几何参数、电子排布、结合能等进行计算,发现CuMAl-LDHs的化学稳定性为CuMgAl-LDHs>CuZnAl-LDHs>CuNiAl-LDHs,与这3种材料的光催化活性以及带隙正好相反.
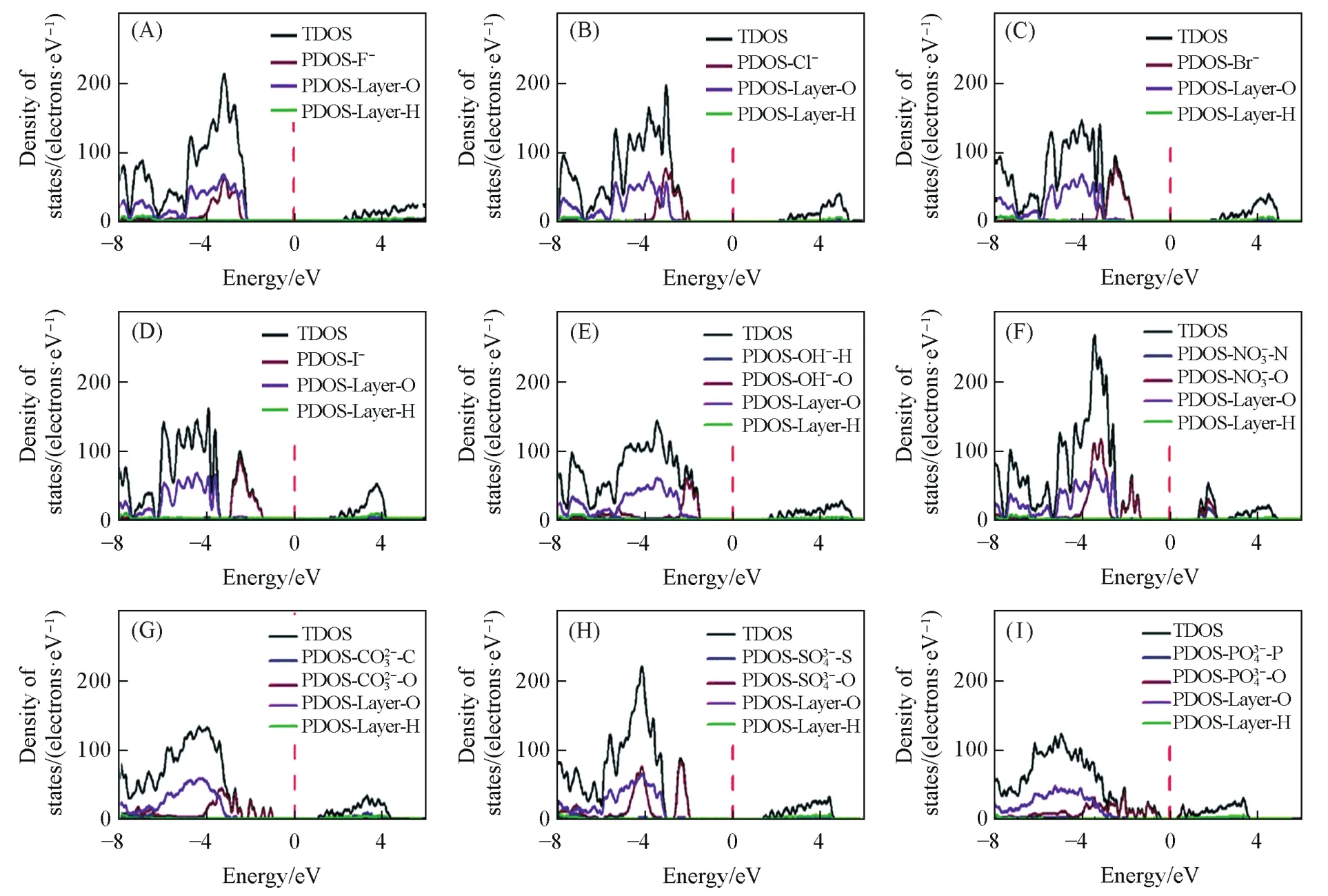
Fig.15 Total density of states(TDOS)and partial density of states(PDOS)for the Mg2Al⁃A⁃LDHs intercalated with different interlayer anions[34]
4.3 碱性位点
已经有大量报道证实以LDHs为前驱体制备的金属氧化物在甲烷化、甲醇合成、高级醇合成和费托化学等反应中表现出良好的催化性能.因此,LDHs的碱性决定了其催化性能[24].而通过理论计算分析态密度(DOS)或分波态密度(PDOS)可以确定酸碱性,通过价带顶距离费米能级的远近可以判断出最碱性位点,这对良好催化性能催化剂的筛选和构筑具有十分重要的意义.Liu等[34]通过态密度分析了MgAl-LDHs中碱性最强位点为层间阴离子,酸性最强位点为层板Mg2+(图15),且阴离子价态与其在价带项(VBM)中与费米能级之间的能量距离是相关的,阴离子价态越高,越靠近费米能级(三价>二价>一价).Leitão等[66]运用DFT方法及GGA+U的交换相关泛函计算了Mg2Fe-NO3-LDHs,Mg2Fe-CO3-LDHs和Mg2Fe-(HPO4)2Na-LDHs的态密度,并分析了价带和导带相对于费米能级的位置得到CO32-的价带更接近费米能级,是碱性最强阴离子.这些酸碱位点和LDHs的催化等性能有关.
电负性和硬度与体系的酸碱性直接相关,LDHs中层板组成元素因其本身性质和在周期表中位置的差异从而使LDHs展现出不同的酸碱性.图16[89]示出了电负性(χ)和硬度(η)与LDHs中二价金属离子有关,且硬度的变化趋势与带隙(HOMO-LUMO带隙[28,29])的变化趋势相同,且HOMO-LUMO带隙越大,硬度越高.含有13族三价阳离子的LDHs比含有过渡金属离子的LDHs更硬,在同一族中,硬度随着周期数的增加而降低,而Fe-LDHs的硬度最低.含有第二主族二价阳离子的LDHs比含有过渡金属阳离子的硬度更强.对于含有13族或过渡金属离子的LDHs,其χ值显示相反的趋势,较高的χ值对应较大的路易斯酸性,因此,含有13族三价阳离子的LDHs比过渡金属离子的LDHs具有更强碱性.在同一族中,碱性随周期数的增加而增加,含闭壳层二价阳离子的LDHs比开壳层过渡金属离子的LDHs碱性更强.这些分析对具有高活性催化材料的构筑和设计具有十分重要的意义.

Fig.16 Mulliken electronegativities(χ)and hardness(η)of M(Ⅱ)M(Ⅲ/Ⅳ)-Cl-LDHs as a function of the atomic numbers of divalent cations[89]
5 结论与展望
研究人员在LDHs的层板组成、层板组成比例、层间阴离子的种类、LDHs层板电荷分布、拓扑结构转变、能带结构、态密度、层间阴离子组成、离子交换性能、主客体作用力、能量性质和光催化性能等方面对LDHs的结构和特性进行了系统的理论研究,揭示了LDHs体系结构-性能间的构效关系,为LDHs或相关材料的设计和制备提供了丰富的理论参考和指导.
但是,由于计算方法的局限性和计算机计算能力等方面的限制,目前LDHs的理论研究仍有很多尚未解决的问题,如更小尺寸以及更大尺寸的LDHs结构和性质有待于进一步完善;不同缺陷类型以及不同缺陷浓度的结构和性质差异以及不同种类阴离子插层LDHs的拉伸和剥离性质还需进一步研究.目前,关于离子交换性质的理论研究大多是在理想条件下进行,溶剂的溶胀性质、交换介质的pH、反应温度、层板组成、层板电荷密度均会对LDHs的离子交换性质产生一定的影响,需要进一步完善.在应用方面,LDHs在光催化、电催化、电子器件、吸附等领域展现出更新更广泛的应用,理论研究仍需要进一步揭示可能的作用机理并用于指导材料的构筑和设计,以及揭示其在各领域中应用的构效关系.随着计算技术的发展,对LDHs材料主客体结构及构效关系的理论研究将会进一步深入,理论研究将会为以LDHs为材料平台构筑一系列基于其超分子插层结构主客体间相互作用的新型功能材料、扩展材料的功能性提供更为丰富和有效的理论指导.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20200404.
猜你喜欢
哈尔滨工业大学学报(2022年5期)2022-04-19
无机材料学报(2020年1期)2020-02-10
纤维复合材料(2018年3期)2018-04-25
浙江大学学报(工学版)(2016年2期)2016-06-05
中国塑料(2016年11期)2016-04-16
中国塑料(2015年6期)2015-11-13
航空材料学报(2015年6期)2015-09-12
现代农业(2015年3期)2015-02-28
应用化工(2014年1期)2014-08-16
郑州大学学报(理学版)(2012年4期)2012-03-25
