
聚氧乙烯基聚合物固态电池的界面研究进展*
2020-12-05 07:34刘玉龙辛明杨丛丽娜谢海明
物理学报 2020年22期
刘玉龙 辛明杨 丛丽娜† 谢海明‡
(东北师范大学化学学院, 国家地方动力电池联合工程实验室, 长春 130024)
聚氧乙烯基聚合物固态电池具有高安全性和高能量密度的特点, 极有可能成为下一代储能器件. 然而,聚氧乙烯基电解质本身的电化学窗口窄, 极大的限制了其能量密度的进一步提升. 目前适配聚氧乙烯基电解质且长循环稳定的正负极材料较少, 这严重阻碍了聚氧乙烯基聚合物固态电池的广泛应用. 其主要问题在于电极材料与聚氧乙烯聚合物电解质之间的负极界面和正极界面都容易发生副反应, 大大地缩短了电池的循环寿命. 为了抑制这些副反应, 人们采取了相应的策略, 取得了一定的成效. 为充分理解固态电池界面处的变化, 可采用各类先进表征手段对其进行研究, 这将为下一步提高固态电池循环稳定性提供更科学的依据.
1 引 言
聚氧乙烯(polyethylene oxide, PEO)基聚合物固态锂电池的发展离不开Michel Armand 教授,正是由于他的开拓和长期坚持才使得这个领域取得了长足的进步[1]. 1980 年, 通过使用PEO-LiSO3CF3电解质, 实现了120 ℃高温下聚合物固态电池的50 次长循环寿命[2]. 随着锂盐、聚合物基体、添加剂和加工工艺的优化, 聚合物固态电池的使用温度逐渐降低到60 ℃. 然而, 由于PEO 类高分子量聚合物的本征性质(融化温度大约60 ℃), 聚合物固态电池的使用温度难以继续降低[3-4].
PEO 基电解质是众多聚合物电解质中研究最早和最多的一类材料. 其导电机理是锂离子与氧化乙烯单元结构上的氧原子不断发生络合和解离反应. 由于PEO 的介电常数低, 这就导致PEO 电解质中离子聚集现象严重, 锂离子迁移困难. 为了减少离子聚集现象的发生, 在聚碳酸酯体系聚合物电解质中引进强极性基团从而提高电导率. PEO 基电解质中的离子迁移主要发生在无定型区域, PEO的融化温度高于60 ℃, 因此其室温电导率低. 相反, 聚硅氧烷基聚合物的玻璃化温度低, 室温下的即可获得高的离子电导率[5]. 此外, PEO 基电解质的电导率与其分子量密切相关. 1993 年, Shi 和Vincent[6]研究了PEO 电解质电导率随聚合物分子量的变化, 分子量范围为0.4 到4000 kg/mol.结果发现PEO 分子量在3200 kg/mol 以下时其离子电导率随分子量的升高迅速降低, 高于3200 kg/mol 之后变化趋于平稳. Teran 等[7]进一步研究了分子量为0.2 到360 kg/mol 的PEO, 实验用的聚合物的分散度更低且高分子量的PEO 样品更多. 结果表明, PEO 电导率随分子量变化的趋势曲线与Shi 和Vincent 的研究类似, 电导率呈指数性降低然后稳定在一个平台.
传统情况下, 聚合物电解质的电化学窗口一般通过测量线性扫描伏安法来获得. 最近, Marchiori 等[8]利用第一性原理对聚合物类电解质的电化学稳定窗口进行了理论计算. 先计算聚合物本身的电化学稳定窗口, 然后将聚合物与不同的锂盐进行搭配来计算电解质的电化学窗口. 根据他们的计算数据,PEO 本身的最低未占据轨道能量是3.13 eV(相对于标准氢电极, 其氧化电位为3.13 V), 最高占据轨道能量是—5.99 eV(相对于标准氢电极, 其还原电位为5.99 V). 这表明PEO 本身的耐氧化性能较差而对锂金属负极的稳定性高(相对于标准氢电极, 锂还原电位为3.65 V). 当PEO 与不同锂盐配合时, 由于氧化过程中聚合物和锂盐结构发生重组从而使聚合物提前发生氧化, 这种结果导致电解质的氧化电位进一步降低. 理论计算结果表明, PEO/LiTFSI 体系的氧化电位是3.57 V(相对于锂金属),这个结果与大部分的实验相符. 为了提高PEO 基聚合物电解质的氧化电位, 需要通过引入譬如碳氮三键、碳氧双键等基团来提高聚合物本身的氧化电位或者通过添加耐氧化的锂盐来抑制其提前分解[8].
目前, PEO 基聚合物电解质的主要挑战是: 室温离子电导率低、电化学窗口窄和界面阻抗高[9].为了解决上述难题, 研究者们提出了不同的策略来提高聚合物电解质的性能. 例如, 为了提高聚合物电解质的电导率, 过去数十年中人们采用各种诸如嵌段共聚物、接枝共聚物、交联共聚物、无机-有机复合、添加剂等策略[10]. 其中, 2005 年报道的纳米结构PEO 固态电解质的室温电导率高达2 × 10—4S/cm, 将其用于LiCoO2/Li 电池中也表现出优异的电化学性能[11]. Tan 等[12]通过在PEO基体中添加多阳离子型离子液体, 增加无定型PEO 比例并促进锂盐的解离, 从而提高了电解质的电导率. Liu 等[13]则利用聚间苯二甲酰间苯二胺(PMIA)作为添加剂, 通过PMIA 和PEO/TFSI之间的氢键相互作用来增加PEO 的无定型比例并最终提高其离子电导率. 该方法所制备的聚合物可与锂金属进行长循环且与LiFePO4正极稳定循环100 圈后保持93%的容量. 为了减少负极界面处锂枝晶的产生, 人们开发了单离子聚合物电解质和其他高离子迁移数体系的电解质, 从而提高负极界面的稳定性[14,15]. PEO 自身对高电压氧化的耐受力较差, 在4 V 左右即发生分解, 因此无法匹配高电压正极材料. 针对该缺点, Zhou 等[16]提出了双层聚合物电解质的构建, 针对正极和负极分别采用不同特性聚合物, 该思想对今后电解质的开发具有指导意义. Liang 等[17]利用腈类电解质碳氮三键的氧化电位高的特点, 将丁二腈与PEO 复合后所制备的固态电解质的电化学窗口提高到4.15 V. 电解质在Li/LiNi0.8Co0.15Al0.05O2(NCA)电池中以1 C倍率循环1000 圈后的容量保持率为66%.
虽然PEO 基聚合物电解质目前存在大量的问题, 但是由于其较高的安全性能仍然受到了广大电池企业和电动车企业的厚爱[18-20]. 法国Bolloré公司联合意大利著名汽车设计公司宾尼法利纳共同出品的Bluecar, 采用高温PEO 体系组装的锂金属聚合物电池, 其能量高达30 kWh, 电动车的最高运行速度为120 km/h, 驾驶距离可至250 km,于2011 年正式进入巴黎汽车租赁市场. 随着人们对高安全性电池的探索, PEO 固态电池的发展也取得了不断的进步, 尤其是对于高电导率和宽电化学窗口的PEO 基聚合物固态电解质的开发[21]. 对于PEO 基聚合物电解质和正、负极的界面研究,最近也获得了大量的关注并取得了重要的进展(图1). 因此, 下面将对PEO 电解质的正负极的界面研究进展展开论述.
2 PEO 基聚合物固态电池的负极界面研究进展
2.1 PEO 基固态电解质与负极界面的反应
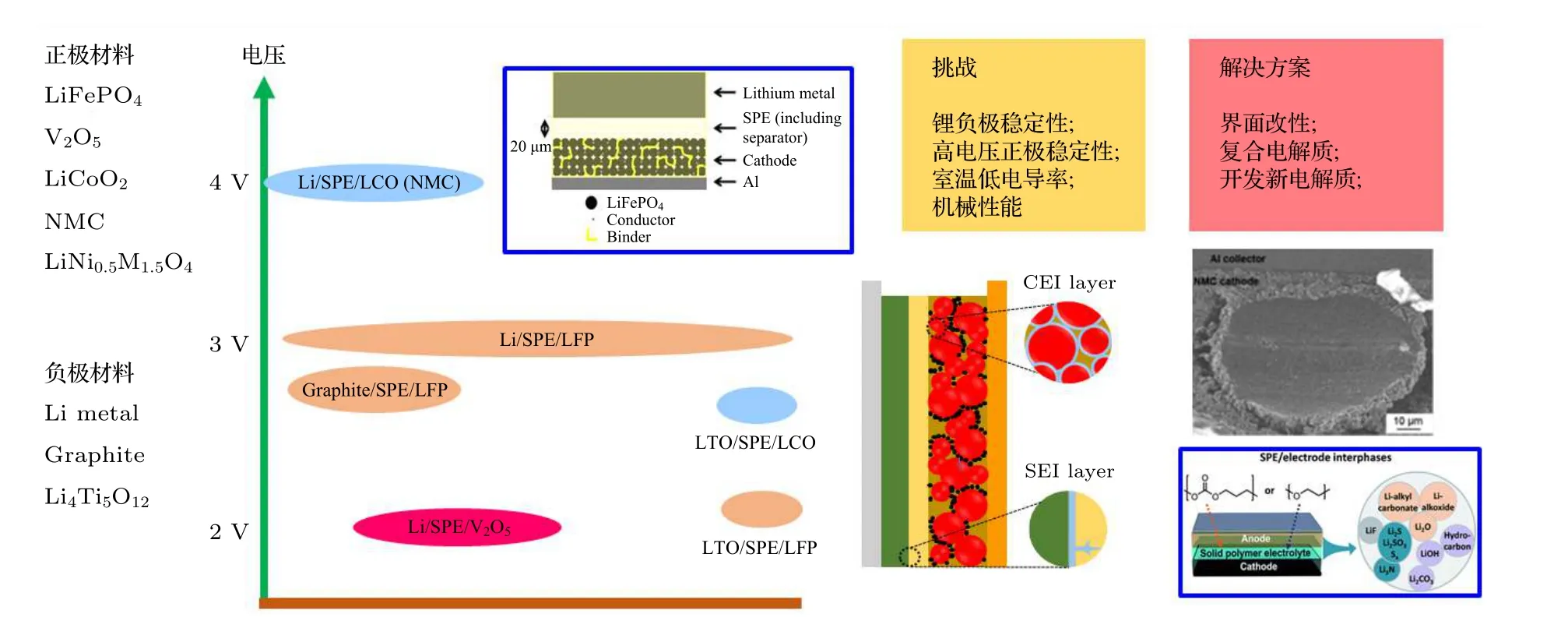
图1 PEO 基聚合物电解质固态电池发展概况.Fig. 1. Overview of the PEO polymer based solid-state battery.
聚合物固态电解质与锂金属的界面反应源于锂金属的强还原性和固态电解质成分的分解.Croce 和 Scorati[22]首次报导了PEO 与锂金属之间的反应, 通过记录Li-PEO 之间的界面电阻随时间的演变, 猜测并证明了锂金属表面有一层钝化膜产生. 然而, 这层钝化膜的存在对于电池性能起到的作用尚无明确定论. Baudry 等[23]研究发现电池在这层钝化膜的作用下仍然可以运行300—400 圈.为了进一步证明聚合物与锂金属之间的界面反应的状况, 人们开始组装锂/聚合物/锂对称电池进行测试. Persi 等[24]利用氧化铝增强的聚合物电解质,通过对称电池测试表明界面电阻在随着时间推移逐渐减少, 60 天以后的界面电阻基本保持稳定, 从而判断通过复合可以减少界面的反应. Li 等[25]利用钛酸钡作为添加剂用于复合聚合物固态电解质中, 结果发现界面电阻随着时间不断的增加, 250 h后仍然不能稳定. 这说明了不同组分的聚合物电解质与锂负极的界面反应不尽相同, 如果对组分进行优化才可以减少界面电阻. Hallinan 等[26]利用X射线光电子能谱(X-ray photon spectroscopy, XPS)研究表明, 产生锂枝晶的锂负极表面有锂盐的分解产物和有机物的分解产物. 利用原子力显微镜(atomic force microscope, AFM)技术和红外光谱手段(Fourier transform infrared spectrometer,FTIR), Mancini 等[27]发现锂金属表面首先形成CF3自由基, 该自由基可夺取聚合物骨架中的氢原子,随后锂原子可能会进一步破坏聚合物中的C—O键形成Li-O-R 型物质. 目前我们对于锂金属和聚合物之间的详细化学反应知之甚少, 在之后的研究中, 需要人们对其进行重点关注.
此外, 由于锂沉积的不均匀性, 聚合物中也不可避免会产生锂枝晶. 聚合物固态电解质中的锂枝晶生长通常与固态电解质的离子迁移数、机械性能以及锂金属杂质有关. 早期的研究表明, 由于聚合物电解质的弹性模量低, 锂枝晶经常在PEO 基的固态电解质中产生. 此外, 聚合物电解质通常在高温下运行, 这会使得聚合物变软, 进一步增加锂枝晶穿透的风险[28]. Harry 等[29]认为, 锂枝晶的生长跟固态电解质表面的不均匀性息息相关. 由于表面杂质的存在, 含有杂质的地方会出现锂的不均匀沉积. 杂质相通常是富氧且绝缘, 这就阻碍了锂金属沉积在其表面. 由于局部电流的升高, 锂的沉积和剥离只能发生在杂质的附近. 此外, 由于不均匀沉积的出现导致杂质上部出现了洞穴. 在两者的同时作用下, 锂金属和固态电解质之间的空隙越来越大, 导致了锂枝晶生长现象加剧(图2(a)). 利用同步辐射X 射线三维成像技术(synchrotron X-ray microtomography, XTM), Maslyn 等[30]发现PSPEO-PS 聚合物电解质中的锂枝晶的生长与电流密度密切相关, 在较低电流密度下, 锂可以发生均匀沉积和剥离, 随着电流密度升高, 界面出现了球形的突出物, 进一步加大电流密度, 除了球状突出物外也出现了锂枝晶. 基于低电流密度下锂的均匀沉积现象, 可以首先在低电流下进行锂的沉积, 得到新鲜的、无杂质的锂薄层. 通过这种方式减少锂表面的杂质含量, 遏制了聚合物与锂金属间的锂枝晶生成进度从而延长了锂金属电池的寿命[31]. Dollé等[32]利用原位扫描电镜技术(in situ scanning electron microscopy, in situ-SEM)观察锂枝晶在PEO中的生长状况(图2(b)). 结果表明随着电流密度的增大, 锂沉积形貌从苔藓状演变成枝晶状, 枝晶生长点位于锂金属与聚合物之间, 严重时枝晶的生长会导致二者剥离进而影响电池的长循环寿命.同时聚合物电解质中锂盐的浓度也会影响锂枝晶的形成. 随着锂盐浓度的增加, 锂枝晶穿透电解质的时间逐渐缩短. 这种现象出现的原因可能是机械强度的变化、界面接触性的变化、电解质电化学性质的变化或者是浓度梯度的变化[33].
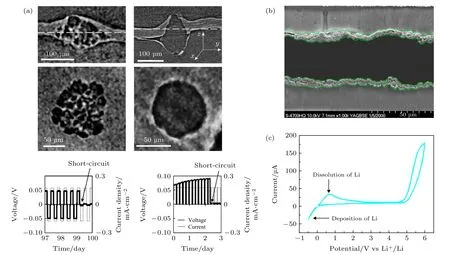
图2 PEO 基固态电解质与负极的反应 (a) 锂的不均匀沉积形成的锂枝晶[29]; (b)锂金属沉积的原位电镜扫描图片[32]; (c) 锂金属沉积的循环伏安曲线[34]Fig. 2. Anode side reaction in PEO based solid state battery: (a) Lithium dendrite formation during deposition[29]; (b) in-situ SEM images of lithium deposition[32]; (c) C-V curves of lithium deposition/dissolution[34].
Golozar 等[35]用扫描电镜技术(scanning electron microscopy, SEM)分析了PEO 固态电池循环后的锂金属形貌和化学成分变化, 结果表明锂金属表面形成了苔藓状和空心针状的锂枝晶. 并且通过化学成分分析发现锂表面形成了锂的碳化物、氧化物和碳氧化物等. 为了理解锂枝晶的生长与聚合物电解质的本征特性的关系, Barai 等[36]通过理论计算获得了聚合物固态电解质的发展方向. 计算结果表明, 制备的聚合物必须具有足够高的弹性模量来减少锂枝晶的形成: 一方面, 通过提高剪切应力来降低尖端处的交换电流密度从而减少锂枝晶的产生; 另一方面, 当聚合物电解质的剪切模量大于锂枝晶弹性模量的千分之一时, 锂枝晶可生长的高度就会由于塑性形变而受到限制. 今后的工作中,人们可以依据该理论指导合成相应的高弹性模量聚合物电解质并对锂枝晶穿透性能进行验证.
最近, Galluzzo 等[37]通过高温下的核磁共振谱(nuclear magnetic resonance , NMR)表征发现锂原子会大量溶解在纯PEO 聚合物当中. 实验中将锂金属/PEO(salt-free)/锂金属对称电池在高温下进行静置, 测试发现整体电池电阻值发生变化,这是因为锂原子溶解在PEO 基体中. 这个现象也能解释和证明聚合物锂金属固态电池中PEO 和锂金属之间的强粘附作用, 同时为PEO 基聚合物电解质的发展提供了很好的指导作用.
除了金属锂负极, 石墨负极也被用于聚合物固态电池的研究中. Zaghi[38]分别采用石墨和钛酸锂用作聚合物固态电池中的负极, 结果发现石墨负极在PEO 基聚合物中的容量衰减很快而钛酸锂负极中没有出现明显的容量衰减. 为了减少石墨负极表面界面反应, Imanishi 等[39]采用PVC 碳包覆的方法改善石墨表面, 提高了石墨负极的充放电容量.为了进一步提高负极的容量, Kobayashi 等[40]也采用了硅碳复合的策略用于PEO 聚合物固态电池中, 最终获得了1000 mAh/g 的可逆容量. 最理想的状况是将高容量的Si 负极用于PEO 电解质中,通过把Si 负极埋伏在碳纸的孔道结构实现与正极材料的匹配, 得到一个高容量的固态电池[41]. 此外,PEO 基聚合物与Na 金属负极之间也有界面反应,Xu 等[42]通过PEO-Na3PS4复合有效地遏制了界面副反应. 虽然其他负极材料也可适用于PEO 基固态电池测试, 但是目前的研究基础还很薄弱, 需要后续的研究对其进一步验证.
2.2 负极界面改善策略
原则上, 可以通过改善锂金属表面SEI 膜的组分和构成来缓和锂金属与PEO 电解质的反应. 其中Appetecchi 等[43-44]发现通过热压无溶剂过程制备PEO 的电解质可以减少负极界面反应.他们认为这样制备的聚合物杂质含量少, 可减少锂金属与锂盐的副反应. Bac 等[45]的工作证实了这个策略的科学性, 他们发现随着锂盐浓度的增加, 负极界面的阻抗会相应地发生变化. 进一步工作发现,用LiFSI 部分替代LiTFSI 可以进一步减少界面的阻抗值. 为了减少界面的阻抗, 也有优先对锂金属表面进行处理形成稳定的界面层, 从而减少界面的副反应[46]. Zeng 等[47]在锂金属表面紫外光原位聚合得到聚醚丙烯酸酯(ipn-PEA)电解质, 电解质的机械强度(12 GPa)高、锂离子导电率(0.22 mS/cm)高. 利用这种电解质包覆的锂金属进行锂对称电池测试, 结果发现沉积的锂形貌十分均匀 (图3 a).
通过构筑无机-有机复合电解质抑制PEO 基聚合物电解质中锂枝晶产生. 引入无机物, 一方面可以提高聚合物电解质的离子电导率, 一方面可以提升机械性能. 电解质本征导电率的升高和机械强度的提高可提升其抑制锂枝晶的能力. 例如, 清华大学Zhao 等[48]将Ta 掺杂的LLZO 颗粒加入到PEO 电解质中, 大大降低了PEO 的结晶温度, 复合电解质的室温导电率为1.12 × 10—5S/cm 且具有良好的机械性能. 同时由于陶瓷颗粒和聚合物基质的相互作用, 阴离子的移动被束缚, 锂沉积的均匀性大大提高. 通过三明治结构的电解质可同时获得高离子电导率和高机械性能, 同时保留聚合物固态电解质的柔性和可加工性能. 这样的设计既可以抑制锂枝晶又能保证锂金属和固态电解质的优良接触. 例如, Huo 等[49]通过设计不同颗粒和体积分数的LLZO-PEO 复合电解质, Li/Li 对称电池在0.2 mA/cm2下稳定循环400 h, 而未出现电池短路现象. 这种设计充分发挥了三明治结构电解质优异的界面接触和锂枝晶抑制的优势(图3(b)).
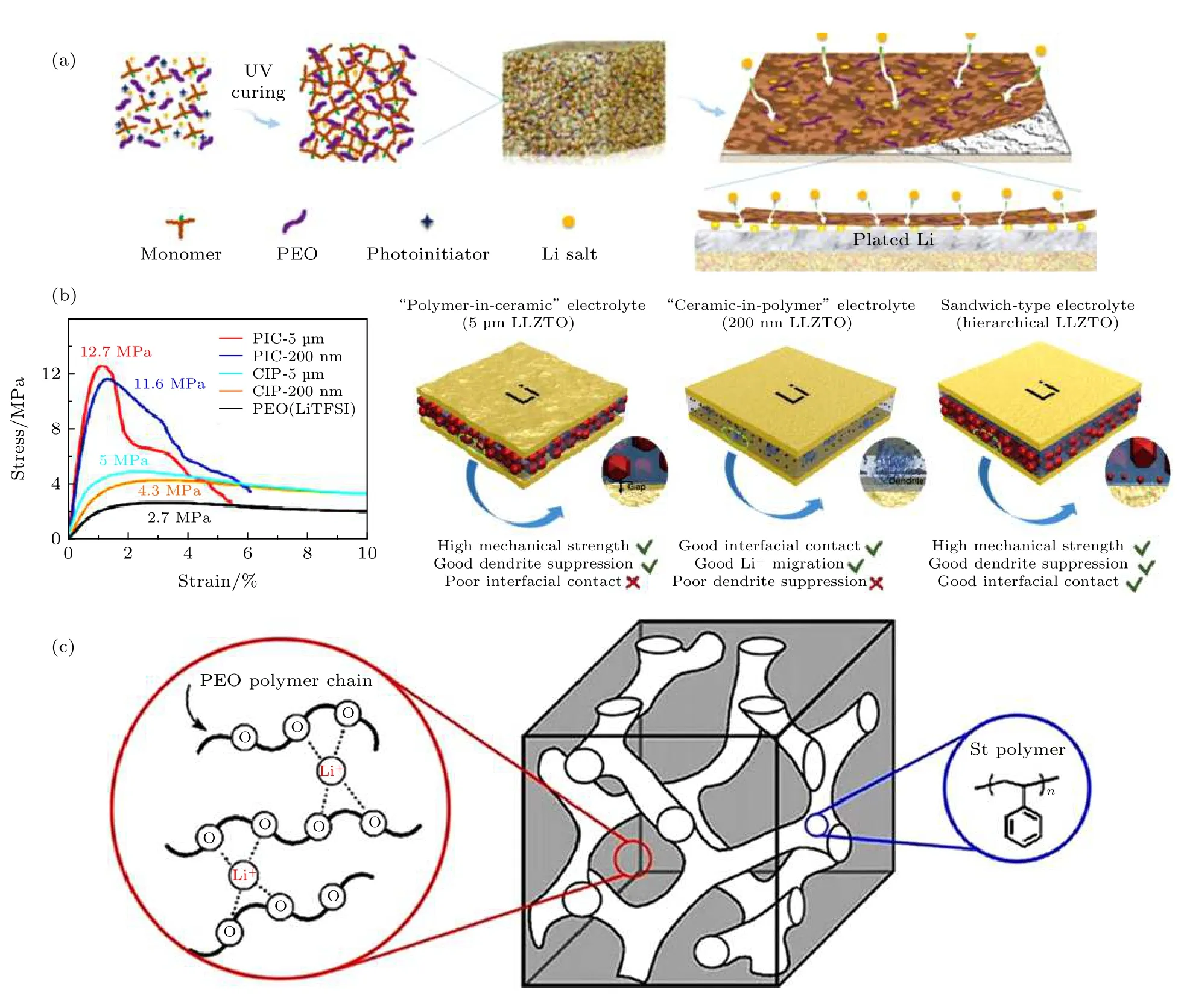
图3 负极界面改善策略 (a) 锂金属表面修饰[47]; (b) 有机-无机复合电解质[49]; (c)嵌段聚合物[54]Fig. 3. Strategies to improve the anode stability[47]: (a) Surface modification of Li metal; (b) organic-inorganic composite electrolyte[49]; (c) triblock co-polymer[54].
理论计算结果表明, 锂枝晶在高弹性模量的聚合物电解质中生长比较缓慢. 通过合成高弹性模量的嵌段共聚物作为电解质的基材可能是一种有效抑制锂枝晶生长的策略. Pawlowska 等[50]、Chintapalli等[51]和Yuan 等[52]制备了以LiTFSI 为Li 盐的PS-PEO SPE 体系, 其结构包含了高模量PS 段及高离子电导的PEO 段. 图3(c)所示是这类嵌段聚合物的示意图, 其中PS 部分用于提高聚合物的机械性能, PEO 部分用于提供离子导电通道. 然而,锂金属表面的杂质可能会导致负极界面出现锂鼓包, 最终引发锂枝晶生长并穿透聚合物电解质. 为了减少这类失效现象, 可以采用先在界面沉积少量锂的预处理方案, 来减少锂枝晶穿透的风险. 实验结果表明, 通过预处理后锂金属在PS-PEO 类聚合物电解质中的循环寿命增加了一个数量级(超过4500 h), 锂鼓包产生的数目也屈指可数[31].
最近, Fu 等[53]提出了其他的策略来抑制锂枝晶的产生. 他们认为可以设计具有与陶瓷电解质类似的低锂偏摩尔体积(partial molar volume of Li+)的“软”离子导体(soft solid-ion conductor )来抑制固态锂金属电池中枝晶的生长. 例如, 通过四丁基氟化铵与锂金属原位生成纳米LiF 并将其限域在刚性聚合物基体(PIM-1)的开放空洞网络中. 然后将LiF@PIM-1 复合物包覆的锂金属用锂电极分部组装技术(LESA)装成扣式电池后进行测试.通过XTM 观察发现, LiF@PIM-1 复合物包覆的锂金属经过160 h 测试后表面依旧平滑而未处理的锂金属负极出现明显锂枝晶. 将LiF@PIM-1 复合物包覆的锂金属组装成锂对称电池后进行长循环、大容量测试, 其寿命长达1000 h.
综上可知, 为了减少负极界面的副反应以及锂枝晶的产生, 可能需要进行多种协同策略从而达到最佳效果. 一方面需要对锂金属表面的物理和化学特性进行修饰实现锂金属的均匀成核, 另一方面通过改善电解质本身的性质来获得高机械性能从而阻断锂枝晶的生长. 或者通过设计低锂离子偏摩尔体积的“软”固态离子导体来阻断锂枝晶的生成.
2.3 PEO 基固态电解质负极界面的表征
为了能更好地理解锂枝晶生长机理并提出合理的策略控制锂枝晶的生长, 对于负极界面的表征是非常重要的. 但是, 直接观测锂枝晶的形成和演化过程是具有挑战性的, 因为锂金属对空气非常敏感且容易被电子辐照破坏. 但经过研究者们几十年的不断探索, 人们还是开发了几种手段对界面进行较为直观的观察.
为了探究锂金属表面与PEO 固态电解质接触前后的变化, Ismai 等[55]应用XPS 发现锂金属表面有LiF 生成. Sun 等[56]研究了PEO 基固态电解质与锂金属界面电化学反应生成的SEI 组分(图4(a)).结果表明, 界面的组分主要由锂盐的分解物如Li2S, Li3N, LiF 组成. 由于PEO 中含有微量的水分, 界面SEI 膜中也探测到了大量的LiOH. Xu 等[57]通过XPS 探究石墨负极表面的SEI 化学组分. 结果发现, 不同于普通的液体电解液, 聚合物固态电解质含有的水分高, 更容易在石墨负极表面产生氢氧化锂并加速锂盐的分解. 这种碱金属氧化物含量高的界面组分是否有益于固态电池的性能尚无定论, 需要人们今后更详细的研究.
SEM 通常是最直观的研究手段, 适用于直接观测PEO/锂金属界面的反应, 特别是锂枝晶的生长状况. Rosso 等[58]利用SEM 结合FIB, 发现界面处的不均匀沉积会形成锂凸起, 这种鼓包最终会导致电池短路. Dollé等[59]利用in-situ SEM 探究了锂枝晶的原位生长过程. 随着电流密度的升高,锂沉积形貌由苔藓状逐渐变成针尖状的锂枝晶. 并且随着枝晶进一步长大, 导致部分位点的电极和聚合物电解质剥离. Hovington 等[60]利用原位截面SEM 分析了两种不同正极的长循环性能并观测了电解质与电极的界面. 结果表明, 采用纳米钒酸锂的正极材料会在正、负极界面处发生聚合物电解质的分解, 从而导致界面层厚度的不断增加. 相反,采用纳米磷酸铁锂正极的界面非常平整, 这可能是因为负极锂表面形成了稳定的SEI 膜. Golozar 等[35]为了探究聚合物固态电解质与负极界面的锂枝晶生长状况, 通过FIB 结合SEM 发现了两种完全不同的锂沉积形貌(图4(b)).
利用XTM, Harry 等[61]分析了聚合物与锂负极界面处的锂沉积情况. 由图4(c)可知, 块状的锂枝晶首先出现在界面之下, 然后逐渐透过聚合物电解质. 通过三维重构技术可以很好的还原锂枝晶穿透的动态过程. 结果证明锂枝晶的生长都发端于界面处而不是电解质内部, 这表明了锂金属表面的性质对控制锂枝晶生长的重要性. 利用XTM 技术去追踪聚合物与锂负极界面的杂质, 可得知杂质相是导致锂鼓泡产生的重要原因. 因此, 实际应用中需要对锂金属进行除杂或修饰, 从而提高锂金属负极的循环寿命[29].
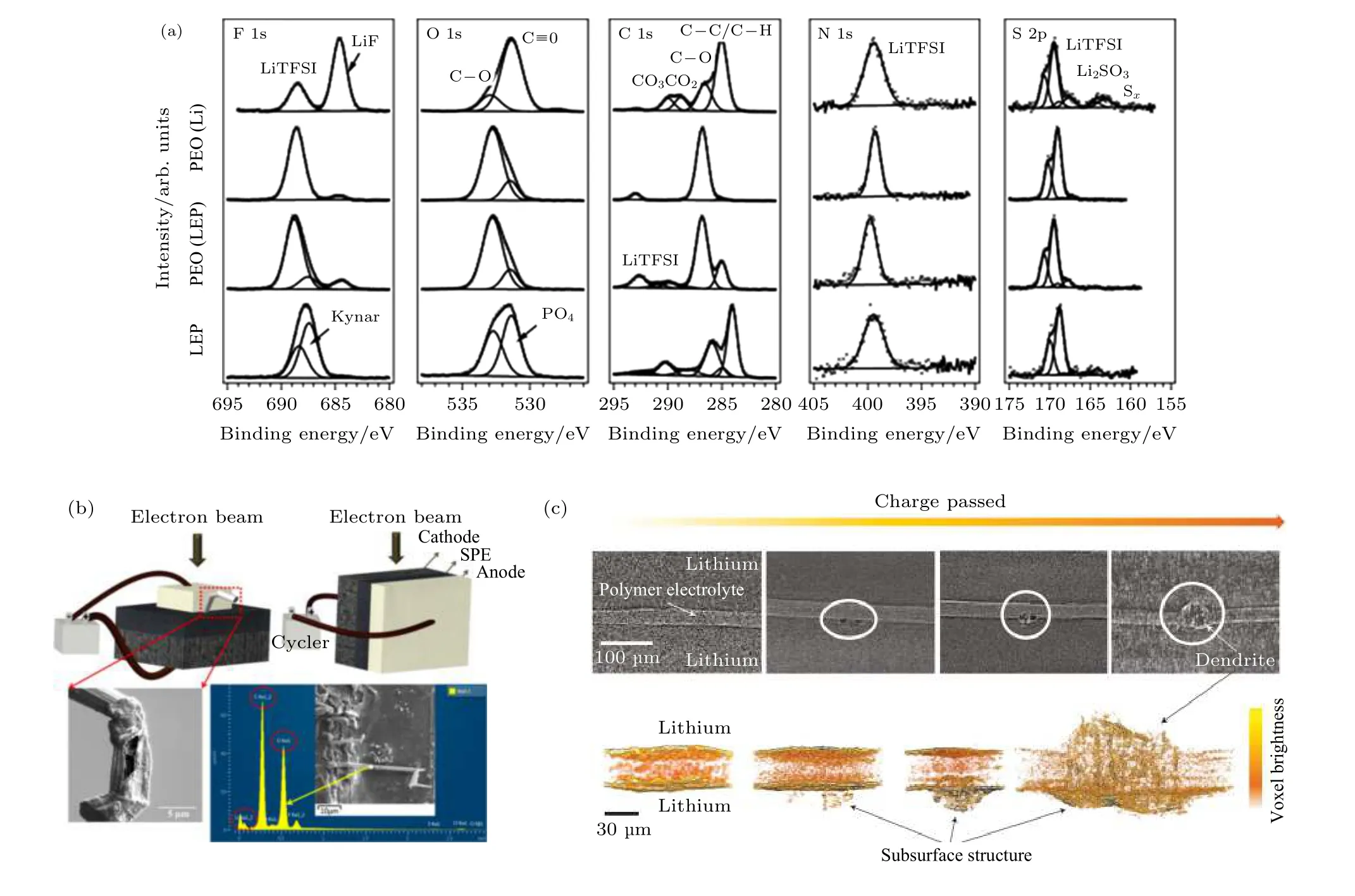
图4 负极界面表征 (a)常规XPS[56]; (b)原位SEM[35]; (c)同步辐射CT[61]Fig. 4. Advanced characterization of anode interface: (a)Lab-XPS[56]; (b)in-situ SEM[35]; (c) synchrotron X-ray microtomography[61].
综上可知, 聚合物电解质与锂金属界面处的微观变化具有复杂性的特点, 需要通过综合研究手段达到科学的结论.
3 PEO 基聚合物固态电池的正极界面研究进展
3.1 PEO 基固态电解质与正极的界面行为
目前PEO 基聚合物固态电解质在提升室温离子电导率, 锂离子迁移数, 机械强度及与金属锂负极兼容性等方面进行了大量的研究工作, 并且均取得了突破性的进展. 然而, PEO 体系在高电压下的热力学和动力学不稳定问题则较少受到关注. 尽管基于PEO 的聚合物电解质与LiFePO4正极具有良好的化学和电化学稳定性, 但有限的理论能量密度限制了固态电池在更高能量密度需求的领域的应用[5,62,63]. 例如, 法国博罗雷研发的以Li|PEO|LiFePO4固态电池组装的“Bluecar”电动车的能量密度仅为100 W·h·kg-1, 远低于普通液体电池的能量密度.
层状过渡金属氧化物正极, 如LiCoO2和LiNixCoyMnzO2, 具有高充、放电电压平台和高的理论比容量(270 mA·h·g—1), 在高能量密度固态锂电池的应用领域表现出极大的潜力[64-66]. 如图5 所示, 以高镍三元材料为正极, 金属锂(Li)作为负极的固态锂电池的能量密度有望突破350 W·h·kg—1, 能够超越现有商用液态锂离子电池的能量密度[67]. 此外, 从结构稳定性方面考虑, PEO 基聚合物电解质有可能抑制三元正极材料中过渡金属离子溶解, 避免层状结构向尖晶石和岩盐相结构转变, 从而缓解高电压正极材料容量衰减和产气等问题[68].
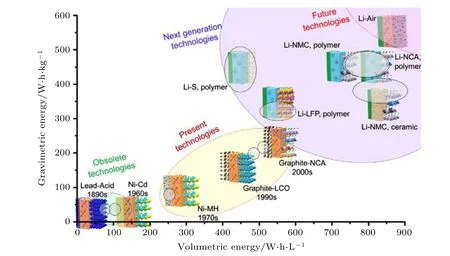
图5 传统电池及全固态锂电池体系质量能量密度和体积能量密度预测与比较图[67]Fig. 5. Prediction of volumetric and gravimetric energy density for traditional battery and all solid-state battery.
另外, 高比容量正极在循环过程中自身产生的体积变化会使界面的接触性变差, 最终导致电池内阻急剧增加[67]. 因此, 为实现更高能量密度、高安全性的高电压聚合物固态锂电池的研发, 现阶段亟需解决高电压正极与PEO 聚合物固态电解质间的界面问题.
3.2 正极界面改善策略
目前, 已有大量的研究工作报道了提升高电压正极与PEO 基聚合物电解质的界面稳定性的方法. 目前采取的改性策略主要围绕两大方面: 1) 正极表面包覆改性; 2) 高电压正极/PEO 基电解质的界面层构建.
3.2.1 正极表面改性
表面包覆改性是最早应用在固态锂电池中解决LiCoO2正极与PEO 聚合物电解质的界面问题的策略[69-77]. Seki 等[74]提出PEO 电解质的氧化分解发生在4.0 V, 连续的氧化分解会增加LiCoO2/PEO 的界面电阻, 导致在4.4 V 截止电压下的循环性能较差. 此外, LiCoO2在持续充电时, 高度氧化的Co4+离子会加速PEO 电解质的氧化分解[73].Yang 等[75]最早采用金属氧化物或磷酸盐作为包覆层材料, 如Al2O3, ZrO2, MgO 和AlPO4, 用于提高LiCoO2的容量保持率和结构稳定性. Miyashiro 等[73]采用喷雾包覆方法将厚度为100 nm 的Al2O3均匀地包覆在正极LiCoO2颗粒表面. Al2O3在锂化过程中生成了少量的LiAlO2, 能够支持界面进行氧化还原反应. 实验结果表明高氧化态的Co4+与PEO 电解质接触面减少, 正极/电解质界面副反应得到一定程度的控制. 然而, 惰性氧化物通常既是电子绝缘体又是较差的离子导体, 会额外增加锂离子扩散阻力和界面电荷转移阻抗. 为了解决这一问题, 大量研究工作采用快离子导体如Li3PO4[74]和Li1+xAlxTi2—x(PO4)3(LATP)[69,70,75-77]作为包覆材料来改善LiCoO2/PEO 的界面稳定性.其 中, NASICON 型Li1+xAlxTi2—x(PO4)3(LATP)固态电解质由于其具有高的离子电导率, 高耐氧化分解电位及低廉的成本而吸引了众多研究者的关注. Morimoto 等[76]通过简单的固相混合方法将Li1.3Al0.3Ti1.7(PO4)3(c-LATP)包覆到LiCoO2表面. 电化学性能结果表明, 5 wt.% c-LATP 包覆的LiCoO2在截止电压为4.5 V 下表现出最佳的循环稳定性. 然而, 固相法制备的c-LATP 颗粒较大,很难保证完全且均匀地覆盖到材料表面(图6(a)).随后, Shim 等[77]采用前驱体包覆方法获得了粒子尺寸相对较小的Li1.3Al0.3Ti1.7(PO4)3, 并用于改性LiCoO2(图6(b)), 在4.5 V 截止电压下, 其循环性能和倍率性能均优于固相法包覆改性的LiCoO2.最近, Li 等[75]采用一种简单易行的溶液方法将0.5 wt% Li1.4Al0.4Ti1.6(PO4)3(LATP)均匀包覆到LiCoO2表面, 获得的材料表面较为光滑, 无明显大颗粒沉积, 包覆层厚度仅为20 nm(图6(c)). 以改性后LiCoO2作为正极、PEO 聚合物作为电解质及金属锂为负极组装的固态电池表现出较高的可逆比容量和更稳定的循环性能. 在充电截止电压为4.2 V, 0.2 C 倍率下进行50 次循环后的容量保持率高达93.2%.
除了高电压LiCoO2正极, LATP 也常作为包覆材料保护高镍三元LiNixCoyMnzO2正极材料[78,79].Wang 等[79]采用简单的机械融合制备Li1.4Al0.4Ti1.6(PO4)3(LATP)包覆LiNi0.6Co0.2Mn0.2O2(NCM 622)正极, 并系统探究了不同融合温度对包覆改性材料电化学性能的影响. 实验结果表明包覆改性后的NCM622 的电化学性能和热稳定性均优于未进行包覆改性的材料. 其中, 经过600 ℃下热处理的 LATP 包覆NCM622, 在4.5 V 充电截止电压下和1C 倍率下循环100 次的容量保持率为90.9%(图6(d)). XPS 结果显示, 随着融合温度的升高,正极表面不纯相Li2CO3的含量逐渐降低, 同时P-O的含量也有所下降, 这说明在高温条件下LATP 存在并逐步扩散到NCM622 表面的趋势(图6(e)—(f)).P, Ti 和Al 等元素适量掺杂到正极结构表面能够有效地抑制高镍材料层状结构的相转变. 因此,LATP 包覆NCM622 电极在高电压下的循环稳定性得到了显著提升.
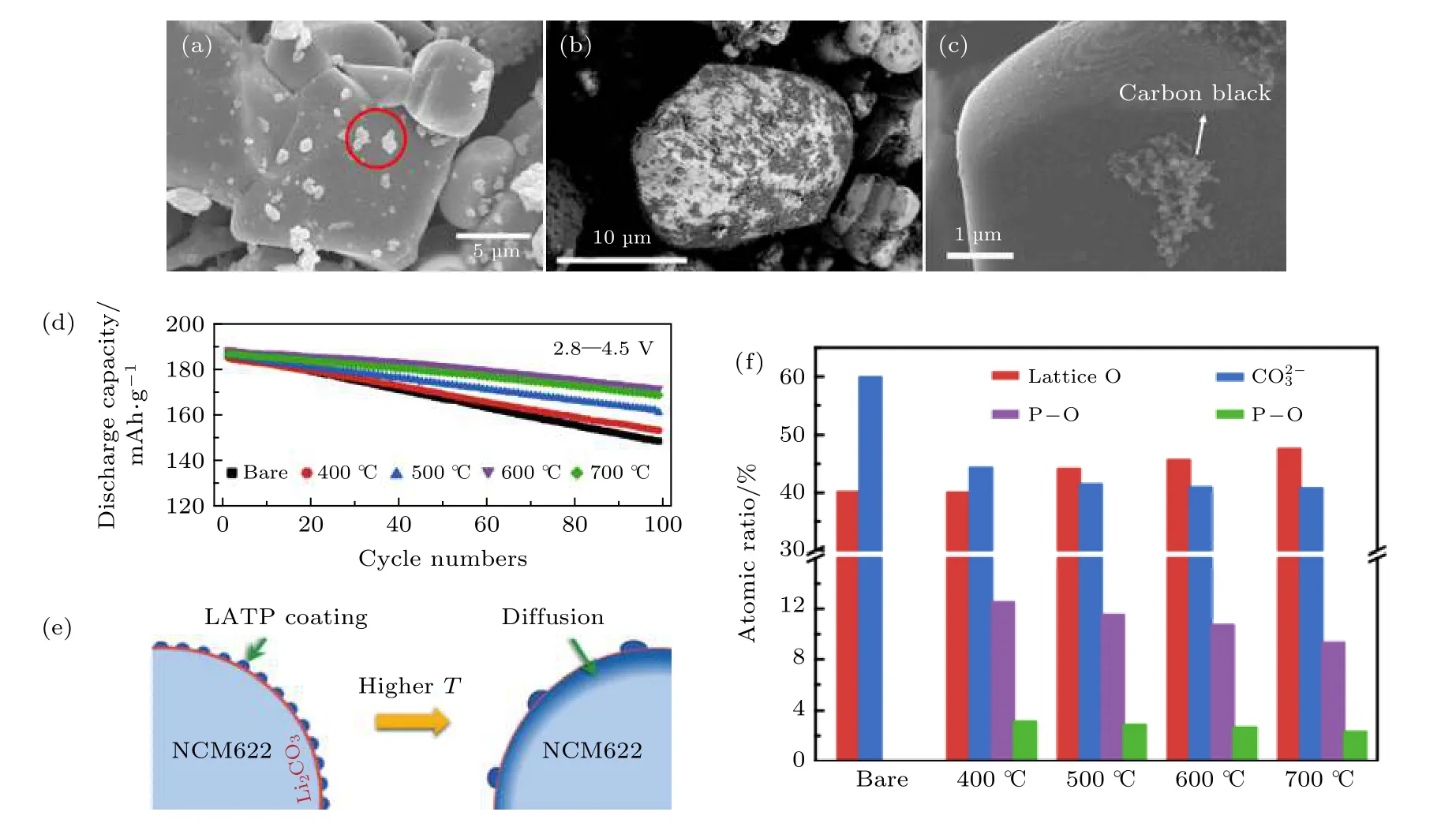
图6 正极表面改性 (a) 固相法制备的5 wt.% c-LATP 包覆的LiCoO2 的SEM 图片[76]; (b) 前驱体包覆方法获得的Li1.3Al0.3Ti1.7(PO4)3 改性LiCoO2 的SEM 图片[77]; (c) 溶液方法制备的0.5 wt% LATP 均匀包覆的LiCoO2 的SEM 图片[75]; (d)在2.8-4.5 V 截止电压下, LATP 包覆及未包覆改性的NCM622 在1 C 倍率下的循环曲线 (1 C = 190 mA/g)[79]; (e) 高温煅烧过程中LATP 包覆改性及未包覆NCM622 的表面演化示意图[79]; (f) 定量分析LATP 包覆改性及未包覆改性NCM622 的XPS 的O1s 和P2p[79]Fig. 6. Cathode interface engineering: (a) SEM image of 5 wt.% c-LATP coated LiCoO2[76]; (b) SEM image of LATP precursor coated LiCoO2[77]; (c) SEM image of 0.5 wt.% LATP coated LiCoO2 by solution method[75]; (d) charge discharge curve of NMC622 at 1 C rate between 2.8-4.5 V (1 C = 190 mA/g)[79]; (e) schematic diagram of LATP coated and un-coated NMC during high temperature sintering[79]; (f) XPS O1s and P2p of LATP coated and un-coated NMC[79].
上述研究表明包覆过程的热处理温度对正极材料的电化学性能的影响是极其重要的. 然而, 元素在无机包覆层与正极界面的扩散通常是相互的.过渡金属离子如Co3+也会不可避免地游离到包覆层表面, 从而影响层状材料在高度脱锂态下的结构稳定性. 近年来, 原子层(ALD)和分子层沉积(MLD)技术作为先进的气相薄膜沉积技术被认为是解决固态电池界面问题最为理想的涂层技术[80](图7(a)).与其他包覆技术相比, ALD/MLD 具有独特的优势, 包括较低的沉积温度, 原子和分子尺度的沉积及优异的均匀性和一致性. Sun 等[81]采用ALD 技术将铌酸锂(LiNbO3, LNO)沉积到LiNi0.8Co0.1Mn0.1O2(NCM811)表面用于改善NCM811 与PEO基聚合物电解质的界面性能. 第一性原理计算结果预测LNO 在高电压下具有良好的化学、电化学稳定性, 高导锂性能和低电子导电性, 因此是包覆材料较为理想的选择[82,83]. 通过调控ALD 沉积圈数可以精准地控制涂层厚度至纳米级, 而涂层厚度是获得优异电化学性能的关键. 结果表明, 10 nm ALD-LNO 涂层能够阻碍长循环后层状结构降解并且抑制晶格氧的损失, 从而稳定了NMC811 活性材料(图7(b)和图7(c)). 另外, LNO 的涂层其较低的电子电导率能够在一定程度缓解高电压下PEO 基聚合物的化学氧化和电化学分解. 最终, 组装的ALD-LNO 包覆的NCM811 |PEO|Li 固态电池在60 ℃, 0.2 C 倍率下循环200 次后平均库伦效率达99.2%, 明显优于未进行包覆改性的NCM811正极(92.3%)(图7(d)). 除了LNO, 其他离子导体材料如Li4Ti5O12[84], LiTaO3[85], LiNbO3[86], Li2SiO3[87]和Li3PO4[88]也可通过ALD/MLD 沉积技术解决电极与电解质的界面问题.
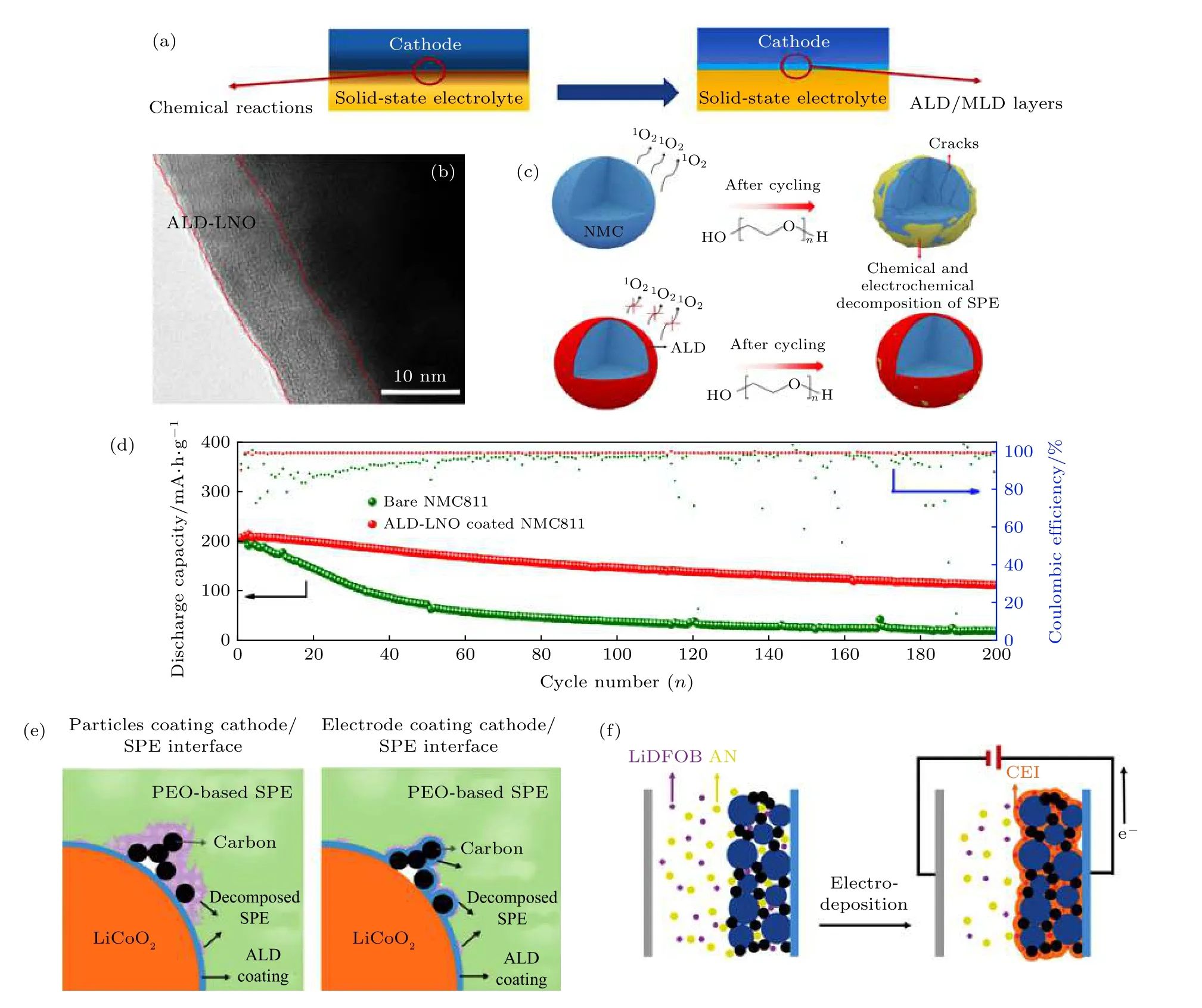
图7 (a) 示意图说明ALD/MLD 技术解决固态电池界面问题[80]; (b) ALD 技术制备的LNO 包覆NCM811 的TEM 图[81]; (c) 示意图说明包覆改性前后的NCM811 正极在长循环后层状结构降解及晶格氧的损失情况[81]; (d) 对比改性前后NCM811 在60 ℃,0.2 C 倍率下循环性能曲线[81]; (e) LTO 包覆LiCoO2 颗粒和LTO 包覆LiCoO2 正极(极片包括活性材料, 导电碳和粘结剂)的示意图[71]; (f) 示意图说明电沉积方法制备PAN 包覆NCM523[91]Fig. 7. (a) Schematic diagram of ALD/MLD in stabilizing cathode interface[80]; (b) TEM image of ALD LNO coated NCM811[81]; (c)illustration of structure degradation and oxygen release of coated and uncoated NCM811[81]; (d) cycling performance of coated and uncoated NCM811 NCM811 at 60 ℃, 0.2 C rate[81]; (e) illustration of ALD LTO coated LiCoO2 particle and LTO coated LiCoO2 electrode[71]; (f) illustration of PAN coated NCM 523 by electrodeposition[91].
目前, 由于其高昂的成本, ALD/MLD 技术不具备广泛适用性. 另外, 高比容量正极在循环过程中产生的体积变化会使无机包覆层开裂或剥离, 从而使包覆层的保护作用失效. Ma 等[72]和Liu 等[89]曾提出采用聚(乙烷基-α-氰基丙烯酸酯)PECA)聚合物作为包覆层改性正极LiCoO2和5 V 高电压正极LiNi0.5Mn1.5O4. ECA 单体能够在室温条件下原位聚合成PECA, 该过程无需高温处理, 并且步骤简单高效. PECA 由于其强吸电子基团-C≡N使其拥有较高的耐氧化能力和快速离子迁移能力.此外, PECA 聚合物包覆层能够阻碍过渡金属离子溶解, 隔绝电解质与电极的直接接触及缓解颗粒微裂现象. 与未改性的固态电池相比, PECA包覆LiCoO2|PEO-LiDFOB|Li 呈现更小的界面阻抗和更优异的高电压循环稳定性[72].
忽然,一只漂亮的鸟儿在殿外跳跃,然后展翅飞远。御座上的孩童对暗潮汹涌的洪流毫无所觉,而是向着飞走的鸟儿,露出了天真的笑容。
先前的大部分研究主要集中在包覆改性正极材料, 往往忽略了导电碳添加剂与PEO 固态电解质的界面稳定性问题. 事实上, Xia 等[90]最早通过循环伏安(C-V)测试结果验证了PEO 与碳材料接触时, 在3.8 V 即开始分解. 因此, 系统地探究包覆改性整个正极极片对界面稳定性的影响, 对揭示界面构建机理是至关重要的. Liang 等[71]对比了钽酸锂(LTO)包覆LiCoO2颗粒和LTO 包覆LiCoO2正极(极片包括活性材料, 导电碳和粘结剂)的电化学性能. 实验结果表明LTO 包覆LiCoO2整个正极在高电压下的循环稳定性更佳. 优异的电化学性能主要受益于LTO 对导电碳/PEO 界面的良好保护, 该研究结果进一步揭示了包覆整个电极对提升正极/电解质界面热力学和动力学稳定性的重要意义. 对于整个电极表面包覆处理实际上是在电极与电解质界面增加一道屏障, 阻碍界Qiu 等[91]提出将前驱体溶液1 M 二氟草酸硼酸锂(LiDFOB)10% 丙烯腈(AN)+EC/DMC 注入到LiNi0.5Co0.3Mn0.2O2(NCM532)极片中, 通过电化学沉积方法原位引发AN 聚合形成PAN 包覆NCM532 电极.这种前驱体溶液电沉积的方式可以确保电极与聚合物包覆层有更强的附着力, 更有利于粒子与粒子之间充分的接触, 在电极内部形成连续快速的离子传导路径. 理论计算结果证实PAN 比PEO 呈现更高的电离电势, 因此具有更高的耐氧化电位. 最终Li|PEO—LiTFSI|NCM523 固态电池在0.5 C 倍率下循环200 次的容量保持率依然可以达到72.3%.Lu 等[92]采用类似的方法, 通过LiDFOB 引发VC原位聚合的方法在LiCoO2表面构筑正极界面层(CEI), 最终该电池可在4.2 V 截止电压下循环500 圈, 容量保持率为71.5%. 并且ARC 测试表明, 固态电池在350 ℃以下不会发生明显的热失控, 证明CEI 膜可同时提升电池的正极界面电化学稳定性和热稳定性.
综合分析, 表面包覆高电压正极能够有效地避免电极与PEO 聚合物的副反应, 降低界面阻抗.然而, 优化包覆层组分仍然是未来应用在固态电池中的一个极大的挑战. 未来重点关注的包覆层材料将以含锂的化合物为主, 筛选的主要依据有: 包覆层材料自身相稳定性、电化学和化学稳定性及离子电导率高低. 除了含锂氧化物, 聚阴离子型氧化物及含硼锂盐作为包覆材料也极具潜力, 未来可应用到高电压固态电池中解决正极/电解质界面问题.
3.2.2 界面层构建
向界面添加缓冲层是解决高电压正极/PEO聚合物电解质界面问题的另一个有效方法. 截止目前, 仍缺乏具有合适的氧化还原电化学窗口的单个聚合物电解质. 通常, PEO 电解质与金属锂相容较好, 但在高压正极侧表现出高的反应性. 而对于一些正极稳定的高氧化电位电解质在负极一侧则表现出高的负极反应性. 为了能够同时满足锂负极无枝晶沉积和高压正极稳定运行的全固态电池[93], Li 和Lucht[94]以及Smart 等[95]设计了一种双层聚合物电解质PEO-LiTFSI/聚N-甲基丙酰胺(PMA)-LiTFSI. PMA 分子含有重复的二甲基乙酰胺(DMAc)单元. DMAc 介电常数高, 能更好地促进盐解离、Li+溶剂化和传输, 另外, DMAc 常用作耐氧化的电解液添加剂. 该PEO—LiTFSI/PMA—LiTFSI 双层电解质能够使Li/LiCoO2全固态电池在4 V 电压下稳定运行, 且经过长期循环后, 也能保持较高的离子电导率. 向正极与PEO基电解质界面添加耐高压界面层的确能够改善过渡金属离子催化PEO 分解反应, 但若想将双层电解质策略广泛应用到未来高电压固态电池基础和产业研究中, 仍需要解决以下问题: 1) 高氧化电位聚合物层的筛选. 高氧化电位聚合物层需要兼具与高电压正极和PEO 良好的兼容性、快速导锂特性和优异的机械韧性; 2) 双层电解质膜的厚度. 未来若要提高能量密度到350 W·h·kg—1高电压固态锂电池, 就要求聚合物电解质膜的厚度低于30 µm.这是目前双层电解质较难突破的技术; 3) 离子在双层聚合物界面传导能力. 双层电解质的应用无疑是在固态电池体系中增加了新的界面, 界面物理接触是否良好, 离子传导是否连续, 是双层电解质能否应用于固态电池中的关键.
除了直接引入界面层, 另一种较为常见的界面构建方式是通过化学和电化学过程在电极表面原位生成界面层. 与直接引入界面层的方式相比, 原位构建的稳定界面层不仅与正极和电解质均能呈现良好的接触性, 而且形成的界面膜的厚度可精确调控.
Qiu 等[96]对PEO 聚合物电解质与LiCoO2正极材料在高电压电化学循环下的失效机理进行了研究. 他们发现在进行4.2 V 恒电位阻抗测试时,其阻抗值随时间增加缓慢, 这表明原位生成的CEI 组分具备导Li+特性且PEO 氧化分解反应在动力学上速率较慢. 然而当匹配LiCoO2正极在4.2 V 进行循环时, 全电池的界面阻抗迅速增大,容量快速衰减. 通过共振非弹性X 射线光谱(RIXS)分析发现, LiCoO2在4.2 V 充电状态时材料中的晶格氧具有强氧化能力, 加速PEO 的氧化分解,而LiCoO2表面失去晶格氧形成尖晶石Co3O4或盐岩相CoO, 均为Li+不良导体, 导致材料的界面阻抗显著增大. 该研究提出如果能抑制正极材料的表面氧化性或者采用表面不具有强氧化性的正极材料, PEO 聚合物电解质有可能在更高的电压下稳定循环. 在实验上也证实了在使用固态电解质LATP 包覆LiCoO2和LiMn0.7Fe0.3PO4正极材料的PEO 全固态电池, 均实现了4.2 V 充电截止电压下的稳定循环. 该研究结果表明在电池体系中电解质的稳定性不仅与材料本身的性质有关, 更与电解质和电极之间的界面特性密切相关. 电极和固态电解质之间的界面设计对最终能否实现可实用化的全固态电池是至关重要的.
此外, 锂盐阴离子能够迁移到电极表面, 并且促进其形成稳定的CEI 或负极界面层(SEI)[97-103].因此, 合理设计锂盐并深入地理解锂盐阴离子分解机制是解决PEO 基聚合物电解质与高电压正极界面问题的有效途径. Zhao 等[104]通过调控聚合物体系的锂盐比例来调节界面性能. 将三锂盐(LiTFSI+5%LiBOB+5%LiNO3)PEO 聚合物电解质组装在Li/LiNi1/3Co1/3Mn1/3O2(NCM111)固态电池中, 在65 ℃和截止电压为4.25 V 条件下,100 次循环后的容量保持率为91.2%, 平均库伦效率达到99%以上. 其中, LiBOB 分解产物主要有BOx, 是CEI 的主要组分, 能够有地效减少正极与电解质的接触面积. Wang 等[105]报道了一种新型的锂硼盐Li[(CF3)3COBF3](LiTFPFB), 并 将 其与PEO 结合构建多层聚合物固态电解质(DSMSPE)(图8(a)). 红外测试结果证实在PEO-LiTFPFB 聚合物电解质中Li+与EO 的络合作用要强于PEO-LiTFSI, 这种分子间的相互作用有利于降低PEO 电解质的HOMO 值. XPS 测试显示循环后的LiCoO2表面界面膜组分为锂硼盐的分解产物,如BOx, LiBxOyFz及LiF 等, 均有利于固态电池获得稳定的界面(图8(b)). 组装的LiCoO2/Li(2.8—4.3 V)电池在60 ℃, 0.1 C 倍率下循环100 次后容量保持率为83.3%(图8(c)).
Li 等[70]认为LAGP 纳米包覆层存在明显的晶粒边界和孔隙, 会导致PEO 的氧化分解, 进而对LiCoO2结构造成损害, 使其近表面晶格结构无序, 从而减缓离子在LiCoO2体相内部的扩散. 因此, 他们提出将LiBOB 调控界面CEI 膜的化学组分策略与LAGP 包覆改性策略相结合, 并应用于改善正极与聚合物的界面稳定性. LiBOB 作为添加剂盐在电极周围的原位分解产物恰好可以填充LAGP 包覆层的间隙, 进一步降低了PEO 氧化分解的可能性. 另外, Fu 等[78]直接将PEO-LiBOB聚合物引入到正极中构建复合正极. PEO-LiBOB既可以作为理想的粘结剂, 又可以作为离子导体相, 不仅能够形成稳定的CEI 膜来抑制聚合物的氧化分解, 同时还能够避免TFSI—阴离子对铝箔集流体的腐蚀. 与直接加入聚合物相比, 直接加入可原位聚合的前驱体溶液更能降低正极/电解质界面阻抗. Lu 等[92]将LiDFOB 溶解于碳酸亚乙烯酯(VC)中构成聚合前驱体溶液. VC 在热引发剂偶氮二异丁氰(AIBN)的作用下原位聚合形成具有耐高压特性的PVC. PVC 与LiDFOB 的协同作用使LiCoO2正极表面形成一层超薄且均匀、低阻抗的CEI 层(图8(d)和图8(e)). CEI 膜改性的LiCoO2|PEO-SPE|Li 固态电池在3.0—4.2 V 电压范围下, 0.5 C 倍率进行500 次循环后容量保持率为71.5%, 而未进行改性的固态电池在200 次循环后容量仅保留至34.1 mA·h·g—1(图8(f)).
综合以上分析, 原位引发聚合构建CEI 膜是改善高电压正极/PEO 电解质界面最有效的解决办法之一. 然而, 目前该类研究工作报道较少, 仍有诸多问题亟待解决, 如原位制备方法的优化, 聚合前驱液的筛选原则, CEI 膜自身柔韧性及导锂性能和CEI 膜先进表征技术等.
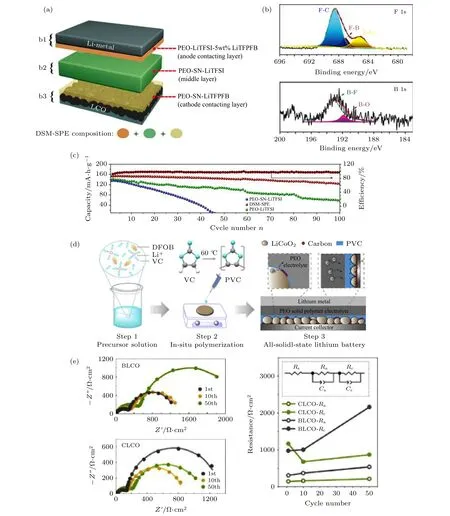
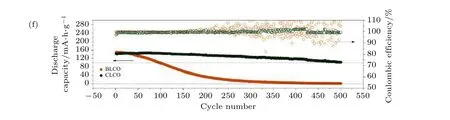
图8 (a) DSM-SPE 电解质膜的结构示意图[105]; (b) DSM-SPE 电解质膜循环10 次后, XPS 测试LiCoO2 正极表面F1s 和B1s 谱图[105]; (c) SPE, DSM-SPE, 和PEO-LiTFSI 电解质组装的LiCoO2/Li 固态电池在2.8-4.3 V 电压范围, 60 ℃, 0.1 C 倍率下循环性能曲线[105]; (d) 原位聚合形成CEI 膜及组装固态电池的示意图[92]; (e) LiCoO2|PEO-SPE|Li 和CEI 膜改性的LiCoO2|PEOSPE|Li 在不同循环圈数放电态的EIS 曲线, 及等效电路图和相应的拟合结果对比图[92]; (f) LiCoO2|PEO-SPE|Li 和CEI 膜改性的LiCoO2|PEO-SPE|Li 在3.0-4.2 V 电压范围, 0.5 C 倍率下的循环性能曲线[92]Fig. 8. (a) Demonstration of DSM-SPE solid electrolyte based solid battery[105];(b) F1s and B1s XPS spectra of LiCoO2 electrode after 10 cycles[105]; (c) cycling performance of LiCoO2/Li cell with SPE, DSM-SPE, and PEO-LiTFSI electrolyte at 60 ℃, 0.1 C rate between 2.8-4.3 V[105]; (d) illustration of in- situ CEI film formation and solid state battery assembly[92]; (e) EIS spectra of LiCoO2|PEO-SPE|Li and CEI modified LiCoO2|PEO-SPE|Li at different cycles[92]; (f) cycling performance of LiCoO2|PEO-SPE|Li and CEI modified LiCoO2|PEO-SPE|Li at 0.5 C rate between 3.0-4.2 V[92].
3.3 PEO 基固态电解质/正极界面反应的先进表征手段
高电压正极/PEO 的界面行为, 包括如界面的动力学、热力学稳定性, 是决定高电压正极固态电池长循环稳定的关键. 然而, 目前对高电压正极固态电池界面的副反应机制和离子传导机理了解尚浅, 仍需先进的原位/非原位光谱和电镜技术协助我们更深入地理解高电压固态锂电池正极界面行为. 迄今为止, 不同的先进表征技术已被报道应用于研究固态电池体系. 这些研究成果对界面行为从不同角度进行剖析, 对未来稳定界面的设计具有重要的指导意义.
3.3.1 微分电化学质谱法
微分电化学质谱法(DEMS)已被广泛用来探究电池体系产气行为, 如锂氧气电池中电化学反应, 锂离子电池中电解液分解反应. 定量分析电化学过程中气体的生成与消耗对于解释电池反应机理、界面反应机理和界面膜形成机理等具有指导意义. Nie 等[69]采用in-situDEMS 技术监测PEO基固态电池在循环过程中气体产生行为. 实验结果表明PEO 聚合物电解质在电压达到4.5 V 时才会明显产生H2. 当PEO 与LiCoO2组装成固态电池,其在4.2 V 就会探测到H2, 这表明LiCoO2与PEO的接触反应会促进固态电池在较低电压下产生H2.他们还通过in-situDEMS 进一步证实Li1.4Al0.4Ti1.6(PO4)3(LATP)包覆改性后能够有效隔绝LiCo O2与PEO 的直接接触, 使产气电位提升到4.5 V.此外, Fu 等[78]通过in-situDEMS 表征手段在循环的PEO-LiTFSI+Super P|PEO-LiTFSI-(Li1.5Al0.5Ge1.5(PO4)3)LAGP |Li 固态电池中探测到CO2(m/z= 44)和O2(m/z= 32)信号, 他们推测, 这些气体也是PEO 在大于4.2 V 电压下氧化分解产生的.
3.3.2 原位/非原位显微镜技术
显微镜技术是一类非常强大的用于研究电极结构和微观界面形貌的技术手段. 在固态电池中用于监测循环过程中正极结构及界面在原子级和纳米级尺度下的演化过程. 例如, Shim 等[77]通过不同的电镜技术分析LATP 包覆层形貌及其对正极LiCoO2微观结构的影响. 由透射电镜(transmission electron spectroscope, TEM)可看出LATP 包覆层呈连续且厚度在0.2 µm(图9(a)). 与之对应的选区电子衍射(selected area electron diffraction, SAED)确定包覆层为NASICON 型的LATP. 不同元素的能量色散谱(energy dispersive spectroscopy, EDS)图显示Co 元素主要存在于正极区, 而Al 元素主要存在于包覆层区域(图9(b)). 另外, Mg 和Ti 元素在两个区域均有分布, 该结果表明LiCoO2结构中有Mg 掺杂, 并且包覆层LATP 中的Ti 元素在高温煅烧条件下会扩散到正极结构中. 为了明确Ti 和Mg 元素在LiCoO2晶格结构中的占位, 研究者采用扫描透射电子显微镜-电子能量损失谱(scanning transmission electron microscope-electron energy loss spectroscopy, STEM-EELS)进行分析, 结果证实少量的Co 位被Ti 元素所取代(图9(c)). 此外, 他们还通过导电原子力显微镜(conducting atomic force microscope, C-AFM)证实Mg 扩散到LATP 层提高了包覆层的电子电导率, 使界面阻抗降低(图9(d)).
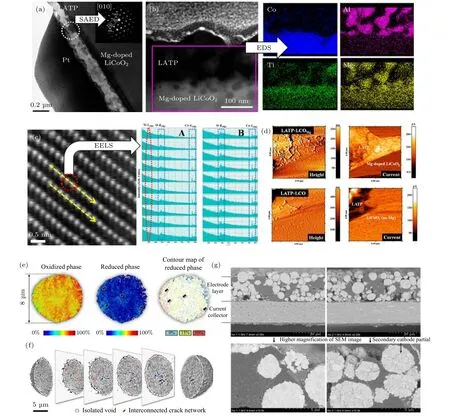
图9 (a) LATP 包覆层 的TEM 和SAED 图[77]; (b) LATP 包 覆 层的STEM 和EDS 图[77]; (c) Mg 掺杂的LiCoO2 的 放 大STEM 和EELS 图[77]; (d) Mg 掺杂的LATP- LiCoO2 和LATP- LiCoO2 的C-AFM 图[77]; (e) 完全充电态的NCA 颗粒在21 周循环后的2DFF-TXM[106]; (f) 完全电态的NCA 颗粒在21 周循环后的3D-FF-TXM[106]; (g) 未进行循环及循环后的NCA 电极在高分辨率下的BIB-SEM 图[106]Fig. 9. (a) TEM and SAED of LATP coating layer[77]; (b) STEM and EDS of LATP coating layer[77]; (c) STEM and EELS of Mg doped LiCoO2[77]; (d) C-AFM of LATP coated Mg-LiCoO2 and LATP- LiCoO2[77]; (e) 2D-FF-TXM of NCA particle after 21 cycle[106]; (f) 3D-FF-TXM of NCA particle after 21 cycle[106]; (g) BIB-SEM of NCA electrode before and after cycling[106].
Doeff 和Liu 等[106]采用先进的同步辐射X 射线全场二维/三维成像技术(2D/3D synchrotron full field X-ray microtomography, 2D/3D-FFXTM)和聚离子束/宽离子束扫描电镜(focused ion beam/broad ion beam-scanning electron microscopy, FIB/BIB-SEM)表征手段观察在PEO基固态电池中循环后的LiNi0.8Co0.15Al0.05O2(NCA)正极材料形貌和荷电状态(SOC)的变化情况. 从图9(e) 2D-FF-TXM 可知NCA 二次颗粒会因循环导致颗粒碎裂和荷电状态(SOC)不均匀. 但考虑到2D-FF-TXM 无法进行离子束方向上的解析,他们又采用3D-FF-TXM 研究了裂纹的3D 进化及其对颗粒内SOC 非均匀分布的影响. 图9(f)的结果直观地呈现了裂纹由颗粒核心向表面传播. 裂纹的增加会引起Li+传输路径弯曲, 最终导致不可逆容量损失和电池阻抗显著增加. BIB-SEM 和FIB-SEM 比2D-FF-TXM 和3D-FF-TXM 拥有更高的空间分辨率, 其得到的结论与前两者结论一致(图9(g)). 由于没有电解液的充分浸润作用,NCA 颗粒的晶间裂痕对固态电池电化学容量和倍率性能的不良影响更为明显.
3.3.3 同步辐射光源光谱技术
同步辐射X-射线吸收光谱(X-ray absorption spectroscopy, XAS)采集时间短, 光源强度可调,对元素价态、未占据电子状态和电荷转移等化学信息非常敏感, 可用于精准判断循环过程中正极界面结构中过渡金属的元素价态和周围电子结构的变化情况. Liang 等[71]对比了未进行包覆改性的LiCoO2在循环前后的XAS 光谱. 循环前后的Co-K 边吸收在全额电子产量(TEY)模式和荧光产量(FLY)模式下基本一致. 这表明LNO 包覆层若只作用在LiCoO2颗粒上, 则对其电化学性能提升的效果并不明显, LNO 完全包覆LiCoO2电极(活性颗粒+导电碳)才能获得明显的性能提升. Li 等[70]将Co K-边光谱与2D TXM XANE 技术相结合分析了包覆及未包覆LAGP 的LiCoO2在3 V 放电状态下Co 价态的变化. 引入2D TXM 技术能够更直观地提供Co 价态变化图像, 方便研究人员清晰地理解材料结构内部价态衍化过程.
同步辐射技术若能广泛应用于研究聚合物固态锂电池体系中原位生成的界面膜组分及其对正极表面晶体结构的影响, 不仅对固态锂电池界面热力学和动力学研究的意义重大, 而且对固态电解质的结构设计及与高电压正极材料的匹配具有指导性作用. 但截至目前, 基于同步辐射技术研究高电压正极与PEO 聚合物固态电解质界面问题的研究结果鲜有报道.
3.3.4 其他重要表征手段
在聚合物固态电池中, XPS 技术常用于分析正极/电解质界面CEI 膜组分[72,78,91,105]. 例如, Ma 等[72]通过XPS 技术分析了LiCoO2|PEO-LiDFOB|Li固态电池正极界面CEI 膜组分. 结果表明CEI 膜主要的组分如LixBOyFz和LiF 是LiDFOB 分解产生的. 除了XPS, 红外光谱技术(FTIR)也常用于确定CEI 组分, 如Qiu 等[91]在循环后的正极表面探测到新的红外峰1397 cm—1, 对应的是LixBOyFz中的B-O 伸缩振动峰, 主要是由LiDFOB 分解形成的. 除了用于表征界面组分, XPS 技术也可用于判断正极中过渡金属元素是否存在溶解和扩散问题.Yang 等[75]通过XPS 的Co2p 谱分析不同LATP 包覆量对Co3+溶解和扩散的影响. 结果显示当LATP包覆量大于0.5 wt%时, 能够完全避免LiCoO2正极在高截止电压和长循环下的Co3+的溶解和扩散.
安全性是聚合物固态电池能否实际应用的一个重要指标. Lu 等[92]采用加速量热法(ARC)测试了LiCoO2|PEO-LTFSI|Li 固态电池的安全性.ARC 测试是在绝热环境中加热电池, 同时监测加热后的热传递反应, 通过跟踪电池热失控期链式反应的动力学可以测量出相关的关键温度参数.
4 结论与展望
本文主要综述了近期固态电池内PEO 基固态电解质与负极、正极界面问题的研究进展. 对于PEO 基固态电解质而言, 与其匹配的正、负极的选择是多样的, 研究的体系也更为复杂. 因此, 分别总结了PEO 基固态电解质与不同的负极和正极的界面问题, 如界面反应机理、界面结构/组分、界面机械稳定性、界面动力学和热力学稳定性问题等.进而, 综述了改进各界面问题的方法与策略, 最后还对相应的表征手段和测试方法进行了详细的介绍. 为了获得高比能、高安全的聚合物固态电池,未来对PEO 基聚合物电解质的研究不再只是集中于离子电导率的提升, 提高其界面兼容性是未来本领域研究的重中之重. 尽管多种改性策略均能在一定程度上稳定PEO 基聚合物电解质/电极的界面,但复杂的界面膜组分、影响界面稳定性的因素和详细的界面动力学过程, 仍缺乏系统的理论认识.
在众多的负极材料中, 金属锂负极以其超高的能量密度、超低的电压平台及与PEO 良好的化学稳定性, 使得其极具潜力应用在高比能PEO 基聚合物固态电池中. 然而PEO 体系固有低的锂离子迁移数和较差的机械强度的缺点同样会导致不均匀沉积, 甚至是锂枝晶的形成. 未来对于负极一侧的界面改性应该更倾向于提升PEO 基聚合物固态电解质的锂离子迁移数, 增加膜的机械模量. 此外,设计具有低的锂偏摩尔体积的“软”快离子导体可以促进锂的均匀沉积, 减少锂枝晶的产生.
在正极一侧, 抑制PEO 电解质与高电压正极的界面反应是实现长循环稳定性的关键. 虽然众多的改性方式已应用于改善正极界面问题, 但仍有诸多不足之处. 例如, 现有包覆改性材料很难同时满足相稳定性、电化学和化学稳定性、高离子电导率及良好机械适应性; 原位引入界面保护膜实际是在复杂的电池体系中增加新的界面, 目前对于该界面是否存在空间电荷层效应, 膜厚度及组分是否可调等问题尚不明确. 未来应该积极开发新的界面改性材料, 通过先进的技术表征手段更深入地理解固态电池的界面化学.
除了以上对于已有改性策略的提升, 未来关于PEO 基固态电解质的研究还应关注以下几方面:
1)拓宽PEO 基聚合物固态电解质的电化学稳定窗口. 利用分子轨道理论, 通过分子结构设计或引入特性官能团调控PEO 基聚合物电解质自身的HOMO 和LUMO 能级. 升高LUMO 能级同时降低HOMO 能级能够拓宽PEO 体系的电化学稳定窗口, 使其同时匹配高电压正极和金属锂负极;
2)构建新型固液混合固态电解质, 改善电极界面稳定性及电极内部离子/电子输运. 添加液体助剂优先在正极和负极界面电化学原位形成稳定的SEI 和CEI 膜, 对正、负极界面起到双层保护,协同提升固态电池的长循环稳定性; 但对于液体助剂的筛选及与电极和固态电解质的兼容性问题尚未有研究工作深入探讨;
3)新型锂盐的开发与匹配应用. 从已报道的原位构建稳定界面的文献可知, 锂盐阴离子若能优先于PEO 聚合物进行氧化/还原分解, 可能促进正、负界面形成稳定界面相层. 因此, 未来合理地设计锂盐阴离子结构或是采用多锂盐的方式有望应用于调控固态电池界面性质;
4)原位实时在线表征手段的运用. 尽管目前材料领域表征手段众多, 但能够用来表征界面组分的技术寥寥无几. 这是由于界面膜组分大多表现出对空气及电子束高度的不稳定性; 通常固态电池界面膜的空间尺寸在纳米级别以下, 而且PEO 基聚合物电解质经过循环后很难从电极上拆解; 高温或是电子束辐射会导致离子、原子分布改变进而导致界面组分改变. 因此, 未来需要更多的原位、无损且清晰的技术进行表征和分析, 如冷冻电镜技术(cryo-EM)和同步辐射技术. 此外, 设计可用于原位表征的电化学电池对同步辐射技术的表征也是至关重要的, 良好的装置能够获得足够强的信号,从而原位实时在线表征界面结构和电荷输运;
5)由于界面现象涉及多尺度和多维度的性质变化, 因此需要不同表征手段相结合来进行更深入的探究. 例如, 2D XANES 与TXM 联用可以同时直观的获得电子结构信息和价态变化情况. 此外,理论计算协助实验技术表征能够给出合理的实验现象的分析和机理解释. 例如, 计算聚合物分子的HOMO 和LUMO 能级判断优先分解的能力; 通过DFT 方法按照不同材料电子隧穿厚度预测CEI/SEI 厚度; 计算聚合物分子与Li+络合数及解络合能力, 并分析Li+在聚合物体系中的动力学行为;计算PEO 在不同活性电极上电化学氧化反应的反应能, 可以判断界面副反应发生的可能性;
6)对于未来PEO 基聚合物固态电池的产业化应用, 首要解决的是高内阻问题. PEO 基固态电池的内阻是等容量液态锂离子电池内阻的100 倍以上, 高的内阻会影响循环性能和电池容量的发挥. 高内阻除了源于电解质自身相对低的离子电导率之外, 界面离子传导也是产生高内阻的主要因素. 因此, 未来界面离子传导动力学的研究有望加速实现PEO 基聚合物固态电池大规模产业化应用.
猜你喜欢
消费电子(2022年6期)2022-08-25
材料研究与应用(2022年2期)2022-05-23
铜业工程(2022年1期)2022-04-13
储能科学与技术(2022年3期)2022-03-23
储能科学与技术(2022年3期)2022-03-23
汽车工程师(2021年12期)2022-01-18
陶瓷学报(2021年1期)2021-04-13
陶瓷学报(2021年1期)2021-04-13
电动工具(2020年2期)2020-04-22
科学与技术(2019年13期)2019-04-19
