
电解液及其界面电化学性质的机理研究进展*
2020-12-05 07:34邢丽丹谢启明李伟善
物理学报 2020年22期
邢丽丹 谢启明 李伟善
(华南师范大学化学学院, 广州 510006)
电解液在锂离子电池中不仅起到传导离子的作用, 其在电极界面所形成的电解液薄层在很大程度上决定了电极/电解液界面的性质, 进而影响电池的循环稳定性、倍率性能和安全性能. 锂离子电池得以成功商品化并广泛应用至今, 与电解液在电极表面分解形成的固体电解质界面膜息息相关. 本文简要综述界面电解液的电化学稳定性、分解规律和影响因素, 旨在抛砖引玉, 引起更多科学家对电解液及其界面性质研究的关注和重视.
1 引 言
2019 年诺贝尔化学奖授予从事锂离子电池研究的三位科学家, 包括约翰·古迪纳夫(John B.Goodenough)、斯坦利·惠廷厄姆(M. Stanley Whittingham)和吉野彰(Akira Yoshino), 以表彰他们在锂离子电池研究领域所作的突出贡献. 锂离子电池作为能源储存器件推动了人类社会能源的变革与创新. 商用锂离子电池一般由含锂过渡金属氧化物正极、石墨负极、碳酸酯基电解液和聚合物隔膜所组成[1]. 锂离子在正负极材料之间可逆脱/嵌, 实现能量的储存与释放. 因此, 锂离子电池的能量密度理论上由正、负极材料决定. 然而, 在实际应用中, 锂离子电池的能量密度能否正常发挥, 以及发挥的稳定性与电解液及其形成的电极/电解液界面性质息息相关[2,3].
上个世纪五十年代, 法国科学家便通过化学反应方法, 发现石墨可以嵌入锂离子并形成石墨层间化合物(GICs), 表明石墨可以作为锂离子电池的理想负极材料[4]. 然而, 当时应用广泛的碳酸丙烯酯(PC)基电解液在石墨电极表面发生持续还原分解(~0.7 V vs. Li/Li+), 导致石墨电极无法到达嵌锂电位(~0.01 V vs. Li/Li+). 直到九十年代初, 科学家偶然发现, 采用PC 的同系物, 碳酸乙烯酯(EC)基电解液, 它可以通过在石墨电极表面发生还原分解, 且还原产物在石墨负极表面形成一层电子不导、离子可导的不溶物质, 从而有效地钝化石墨负极, 抑制电解液的后续还原分解, 使得锂离子可以在石墨负极中可逆嵌入/脱出. 这层钝化层被称之为“固体电解质界面膜”, 简称SEI 膜. 至此,第一代以石墨为负极的商品化锂离子电池才得以问世并应用至今.
最初商品化的钴酸锂/石墨电池工作电压只有4.2 V, 电池充电截止电压尚未超过碳酸酯基电解液的氧化分解电位(4.3 V), 因此仅需要关注石墨负极界面SEI 膜的性质及对电池性能的影响(如图1, 电池2). 然而, 随着高能量密度锂离子电池的发展, 各类高比容量和高工作电位的新型正极材料, 以及高比容量负极材料(例如金属锂, 硅等)应运而生. 由这些材料构成的高能量密度锂离子电池工作电压已远远超出碳酸酯基电解液的氧化稳定性; 而且, 高比容量的负极在嵌脱锂过程体积膨胀/收缩较为严重. 另外, 对于锂金属电池(以金属锂为负极)而言, 还存在负极锂枝晶增长的安全问题[5]. 因此, 这些新电极材料对电池正、负极界面的电化学稳定性和结构稳定性提出了更高的要求(如图1, 电池4). 寻找开发具有更宽电化学稳定窗口(在电池的工作电压范围内, 电解液不发生电化学分解)的新型电解液是解决这些界面不稳定问题的根本方案. 与此同时, 通过在现有碳酸酯基电解液中引入高效的成膜添加剂, 在高电压正极和高比容量负极表面分别构筑稳定的CEI(正极固体电解质界面)膜和SEI 膜, 也有望突破下一代锂离子电池的应用瓶颈.

图1 锂离子电池的工作电压窗口与SEI/CEI 膜的关系Fig. 1. Relationship between electrochemical window and SEI/CEI film of lithium-ion batteries.
由于固体电解质界面膜主要是电解液中的某些组分在电池首次充电过程中, 在正/负极表面发生氧化/还原分解原位形成的. 参与分解的电解液组分复杂且处于动态演变过程, 难以确定; 其次,所形成的界面膜组分对环境高度敏感, 电池体系高度密封, 难以开展原位研究, 导致人们对SEI/CEI膜的形成机制的仍存在许多的问题[6,7], 例如, 哪些电解液组分参与成膜反应, 如何分解, 受哪些因素影响等. 针对这一研究现状, 本文主要对锂离子电池电解液/成膜添加剂在电极表面发生电化学分解的规律和影响因素的相关研究进行简要综述, 旨在抛砖引玉, 引起更多科学家对电解液及其界面性质研究的关注和重视.
2 界面电解液组分与本体电解液组分比例的差异
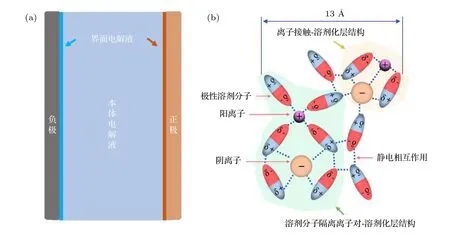
图2 (a) 电池中界面电解液与本体电解液的差异; (b) 界面电解液的主要结构Fig. 2. (a) Difference between the interfacial electrolyte and the bulk electrolyte; (b) main structure of the interfacial electrolyte.
目前应用最广泛的锂离子电池电解液主要是由六氟磷酸锂(LiPF6)盐溶解在环状和链状碳酸酯混合溶剂中所组成. 当电解液加入到电池中的时候, 由于电极材料与电解液中的某一组分可能存在一定的物理/化学特性吸附, 而在电极表面形成一层(大约13 Å厚)的界面电解液层, 如图2(a)所示[8].当电池进行充放电时, 由于正、负极界面开始形成电场, 将进一步促使界面电解液组分与本体电解液组分比例差异的增大. 此时, 决定电极/电解液界面性质的是界面的电解液组分, 而不是本体电解液. 如果界面电解液组分的电化学稳定窗口比电池的工作电压宽, 那么电解液在电池充放电过程中不发生电化学分解, 电极/电解液界面达到真正意义上的稳定; 如果电池的工作电压超出界面电解液的电化学稳定窗口, 且界面电解液的分解产物无法钝化电极表面时, 则电解液会发生持续分解, 导致电池无法进行正常充放电; 如果界面的电解液发生电化学分解后, 产物可以钝化电极表面, 即形成固体电解质界面膜, 则可以使得界面在某种特定的环境和条件(温度、电压、外力)下达到稳定.
界面电解液组分的结构示意图如图2(b)所示.离子与极性溶剂分子之间通过静电作用形成复杂的溶剂化层结构, 主要包括溶剂分子隔离离子对和离子接触溶剂化层结构. 极性的溶剂分子之间也存在一定的静电相互作用. 由此可见, 电解液各组分之间是相互联系, 不存在孤立的组分. 研究界面电解液组分的电化学稳定性和分解机理, 必须考虑组分之间的相互影响. 接下来, 我们将举一些具有代表性的研究例子来说明这一认识的重要性.
量子化学计算方法可以从分子层面研究小分子/离子体系的电化学稳定性, 已广泛用于电解液组分电化学稳定性和分解机理的研究[9-12]. 通过优化研究体系的结构, 可获得其前线分子轨道能量、电子亲合能和电离能, 这些数据可用于判断体系之间的电化学反应活性顺序; 通过计算体系失去/得到电子前后自由能的变化, 结合ΔG= —nEF公式,可以获得体系的理论氧化/还原电位[13], 进而推测发生电化学分解的主要成分. 需要注意的是, 这类计算忽略了电极材料的影响, 因此计算得到的绝对数值(能量或者电位)不能与实验值直接比较. 但是计算得到的不同体系的电化学活性顺序, 可用于预测/判断电解液体系在惰性电极上的反应活性顺序.
锂离子电池电解液通常使用碳酸乙烯酯(EC)和碳酸二甲酯(DMC)混合溶剂. 理论计算结果表明, 不管是否考虑锂盐阴离子的影响, 获得的EC氧化稳定性都比DMC 高, 说明后者比前者在电池充电过程中更容易发生氧化分解[9,14]. 但是, 在电池的实际循环过程中, 实验检测到的电解液分解产物却大部分来自具有较高氧化稳定性的EC[2]. 为了解释这一理论与实验研究结果的差异, 我们推测有可能是由于正极界面电解液中EC 的含量高于DMC, 导致氧化稳定性较高的EC 成为氧化分解的主要组分. 计算结果表明, 电解液中的锂盐阴离子与EC 溶剂分子的结合能力明显强于DMC.而带正电的高电压正极表面会富集阴离子. 这些会携带与其结合能力更强的EC 富集在正极表面[9]. Vatamanu 等[15]随后模拟含EC 和DMC混合溶剂的电解液, 研究电极界面电解液组分与电极电势的关系, 结果如图3 所示. 与我们预测的结果一致, 在高电压正极表面含量高, 且EC 的含量明显高于DMC, 进而解释了EC 成为主要氧化分解组分的原因. 与此同时, 可以注意到在带负电的电极界面上, EC 的含量同样明显高于DMC.EC 这种在带电电极表面富集的行为主要是由于其分子极性比DMC 大, 成为锂离子和阴离子溶剂化层中的主要成分, 在充电过程中伴随离子迁移至电极表面. 而且, 带电的电极表面吸附极性较大的分子更稳定(生成能更负).

图3 (a) 界面电解液中各组分的含量与电极电势的关系; (b) 不同电极电势下的界面电解液图片[15]Fig. 3. (a) Relationship between the component of interfacial electrolyte and the electrode potential; (b) snapshots of interfacial electrolyte at various electrode potential[15].

图4 (a) 界面电解液中TMC:DMC 比例与电极电势的关系; (b) 带正电电极界面电解液中TMS 和DMC 溶剂的微观结构示意图[16]Fig. 4. (a) Ratio of cumulative densities TMS:DMC in the interfacial electrolyte as a function of electrode potential; (b) schematic diagram of the microstructure of TMS and DMC solvents in the electrolyte at the interface of positively charged electrodes[16].
基于这一认识, 我们研究了含环丁砜(TMS)和DMC 混合溶剂的电解液在电极界面形成的组分与电极电势的关系, 结果如图4(a)所示[16]. 即便是在本体电解液中TMS∶DMC = 1∶2 的情况下,TMS 在带负电电极界面的含量仍明显高于DMC(约1.5 倍). 这就解释了在TMS 基电解液中引入具有SEI 成膜功能的共溶剂分子DMC, 却无法解决TMS 与石墨负极相容性差的问题. 另外, 在带正电的电极表面, TMS 的含量与DMC 相当, 接近本体电解液中的比例. 然而, 研究界面层中TMS 和DMC 两种溶剂分子的结构可发现(图4(b)), TMS的=O 基团比DMC 分子的更靠近电极表面, 因此界面性质主要由TMS 决定. 而TMS 的氧化稳定性比DMC 更高, 解释了在具有高氧化稳定性的电解液体系中, 引入氧化稳定性较低的DMC, 却不会降低其高电压电极界面稳定性的实验结果[17].
上述研究结果表明, 界面电解液组分的电化学稳定性才是决定界面性质的关键. 通过比较分子极性大小, 可以初步预测含混合溶剂的电解液在带电电极界面形成的主要成分, 利用这一规律, 可以解释在电解液中引入低极性的溶剂体系, 仅改变电解液的本体宏观物理性质, 而不影响界面性质的实验现象[18]. 更重要的是可以用于指导新型电解液体系的设计及理想界面的构筑.
3 界面电解液组分氧化稳定性的影响因素
电解液中的离子-离子, 离子-溶剂分子, 极性溶剂分子-极性溶剂分子之间均存在静电作用, 如图2(b)所示. 我们前期的研究结果表明, 这些相互作用结构对溶剂分子的电化学稳定性具有不可忽略的影响. 当研究界面电解液组分的电化学稳定性时, 如果仅研究其中的一个孤立组分的稳定性(例如单个溶剂分子/离子), 获得的结果有可能是错的.
以碳酸丙烯酯(PC)为例, PC 分子失去一个电子氧化后, 发生开环反应, 计算得到的氧化电位是7.08 V (vs. Li/Li+, 下同), 见图5. 而当PC 分子与或者形成的离子-溶剂分子结构失去一个电子时, PC 分子结构的变化与单个PC 发生氧化的变化截然不同. 此时, PC 分子上CH 基团H 发生转移, 分别与和结合产生HF 和HClO4, 而且体系的氧化电位由7.08 V 分别下降至6.47 和5.57 V. PC-是所研究的三个体系中氧化电位最低的, 这与实验结果检测到含LiClO4盐的电解液氧化稳定性最差相吻合[13]. 如图5 所示, EC 的氧化稳定性受锂盐阴离子的影响与PC类似[19]. 另外, 当溶剂分子周围存在另一个溶剂分子时(极性溶剂分子间也存在相互作用, 如图2(b)所示), 其氧化稳定性和分解产物也有差异. 例如,EC-EC 结构失去一个电子时, 其中一个EC 分子的-CH2—基团H 转移至另一个EC 分子的羰基氧上, 体系的计算氧化电位由7.01 V 下降5.94 V. 由此可见, 阴离子和共溶剂分子对所研究的溶剂分子氧化稳定性和分解产物均有不可忽略的影响.
寻找开发高氧化稳定性的新型电解液体系是高电压锂离子电池电解液的重点研究方向. 在所报道的电解液体系中, 以丁二腈(SN)为添加剂的体系由于具有优异的氧化稳定性而备受关注. 在碳酸酯基电解液中添加少量的SN, 可以有效地抑制碳酸酯基电解液在Pt 电极上的氧化分解[20,21]. 由于在LSV 测试曲线中观测不到与SN 添加剂相应的氧化峰电流, 而且SN 分子的最高电子占据轨道(HOMO)能量明显低于碳酸酯溶剂分子, 表明SN 失去电子的能力较碳酸酯弱. 因此, 前期的研究普遍认为, SN的耐高电压机理是其通过在高电压电极表面吸附一层单分子SN 层, 这层SN 分子具有高氧化稳定性, 可以在高电压条件下保持稳定, 且抑制碳酸酯基电解液的氧化分解, 即达到了真正意义上的界面稳定的效果.
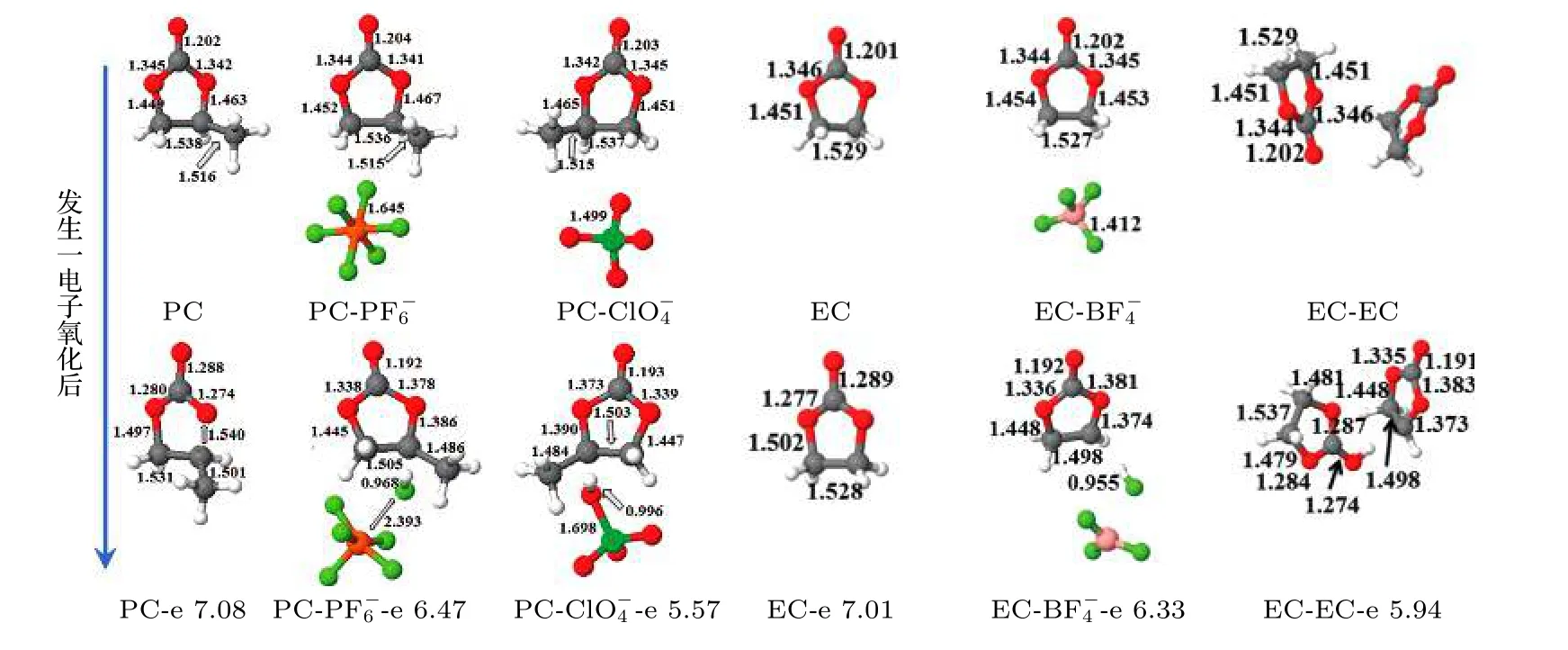
图5 锂盐阴离子和共溶剂分子对PC 和EC 氧化稳定性的影响[13,19]Fig. 5. Effects of anions and co-solvent molecules on the oxidative stability of PC and EC [13,19].
然而, 前面研究结果表明, 不能简单地通过一个孤立的溶剂/添加剂分子的前线分子轨道来判断其在电解液中的电化学稳定性. 其次, 在LSV 测试中没有观测到相应组分的氧化电流, 不一定代表该组分没有发生氧化分解. 若该氧化反应速率较慢,反应不受反应物扩散速度的限制, 则无法观测到相应的反应峰电流. 基于上述的认识, 我们采用计算和实验相结合, 详细研究了含SN 添加剂电解液的耐高电压机理[22]. 图6(a)表明, 单分子体系时, SN 的计算氧化电位确实明显高于碳酸酯分子, 与所报道的HOMO 计算结果一致. 但是, 当考虑以及共溶剂分子的影响时, SN 分子的氧化电位下降比碳酸酯溶剂分子还明显, 且氧化电位最终低于碳酸酯分子体系. 与此同时, 我们设计了一系列的验证实验,如图6(b)所示. 理论计算与实验结果最终证明,SN 在电解液中其实是起到成膜添加剂的作用.SN 优先于碳酸酯基电解液发生氧化分解, 产物在高电压正极表面形成固体电解质界面膜, 从而达到提高高电压电极/电解液界面稳定性的效果(图6(c)).
值得一提的是, 有一部分溶剂的氧化稳定性,如甲基乙基砜, 受锂盐阴离子和共溶剂分子的影响很小, 其电解液的表现出来的电化学氧化稳定性比碳酸酯溶剂体系高[23]. 因此, 耐高电压电解液的开发方向主要可以从两方面着手, 一方面, 就是保留LiPF6盐, 寻找设计具有对稳定的耐高电压溶剂, 如甲基乙基砜一类; 另一方面就是保留碳酸酯溶剂分子, 寻找设计具有高电压稳定的新型锂盐(即对碳酸酯溶剂分子的氧化稳定性没有催化降低作用).
4 界面电解液组分还原稳定性的影响因素
与电解液的氧化稳定性和分解机理类似, 锂盐阴离子对碳酸酯溶剂分子的还原稳定性和分解产物影响非常大. 在研究界面电解液组分的还原分解时, 务必考虑离子溶剂化层中各组分的影响.
界面电解液的组分对负极界面性质影响最典型的例子, 莫过于“PC-EC”两种溶剂分子结构相似(相差一个甲基), 却引起截然不同的石墨负极界面电化学行为. PC 基电解液在石墨充电(电极电势下降)过程中, 在0.7 V 的电位下持续还原分解,导致石墨负极无法下降至更低的嵌锂电位. 而把PC 溶剂改为EC 时, 电解液在0.8 V 发生还原分解, 并形成稳定的SEI 膜钝化石墨负极, 使得锂离子可以在更低的电位下可逆嵌入/脱出石墨负极.前期关于“PC-EC 在石墨负极界面差异”机理的研究仅关注溶剂分子本身的还原稳定性和分解产物[24],或者不含阴离子的离子溶剂化层的体积大小[25],导致所提出的机理仍存在一些问题. 另外, 值得一提的是, SEI 膜的组分除了来自溶剂分子的还原分解产物, 还有一部分来自锂盐的分解产物, 例如LiF和LiPOxFy. 可见,参与形成负极的SEI膜, 只是在前期的机理研究中均被忽略了.
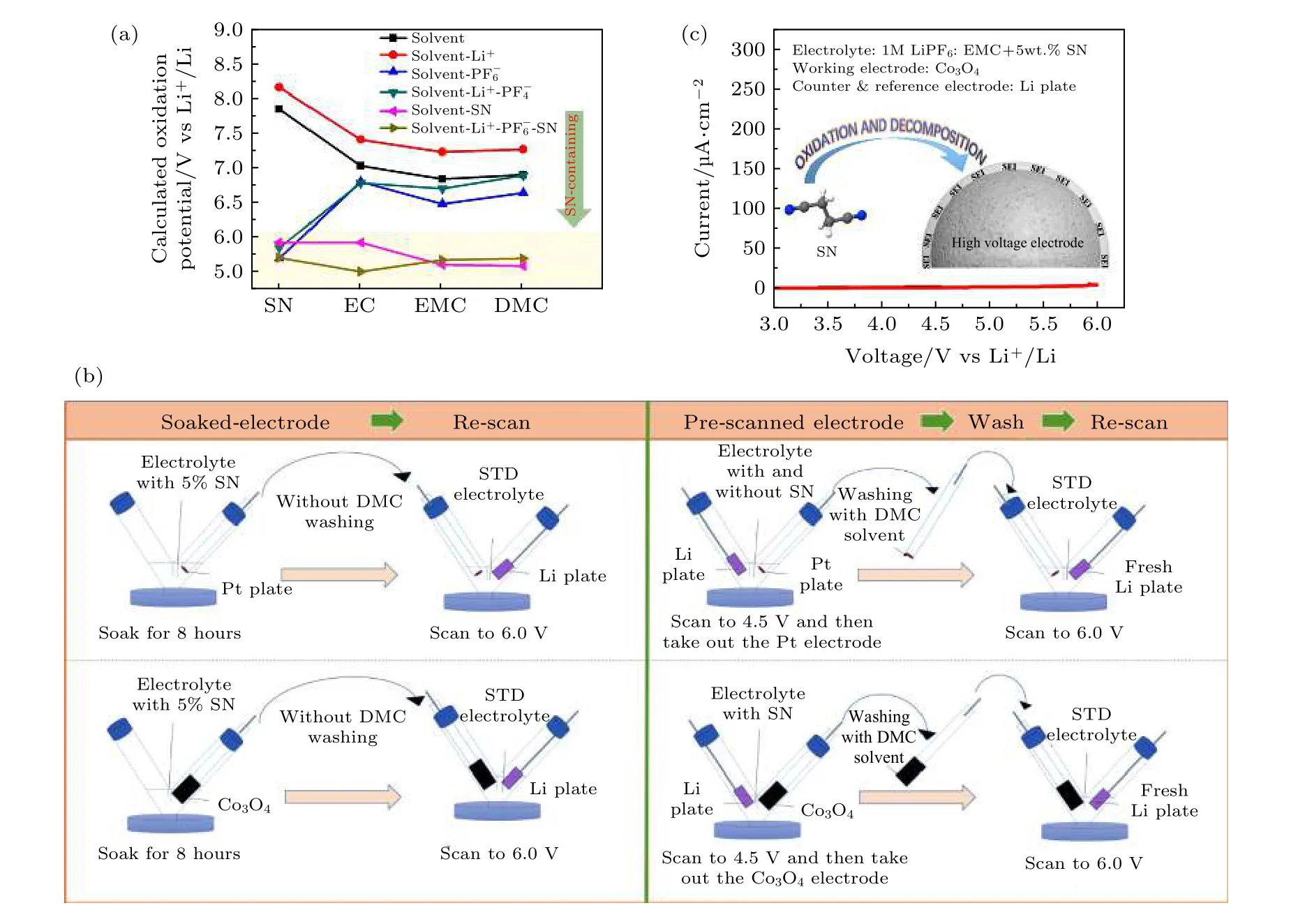
图6 (a) 和共溶剂分子对SN 和碳酸酯计算氧化电位的影响; (b) 不同的实验设计验证SN 添加剂的作用机理; (c) SN 添加剂提高高电压电极/电解液界面稳定性的机理图[22]Fig. 6. (a) Influence of and co-solvent molecules on the calculated oxidation potential of SN and carbonate; (b) different experimental designs verified the mechanism of SN additive: (c) mechanism of SN additive on improving the stability of high voltage electrode/electrolyte interface[22].
基于这一研究现状, 我们详细研究了锂盐LiPF6分别在PC 和EC 溶剂中的解离过程及后续形成的溶剂化层结构[26]. 两种溶剂分子在本体电解液中所形成的溶剂化层结构没有差异. 这些溶剂化层的体积明显大于石墨层的层间距, 如图7(a)所示. 因此, 在电场及石墨层间距位阻双重作用下,锂离子在嵌入石墨层之前必须先脱溶剂化. 我们的计算结果发现, EC 基电解液中, 锂离子溶剂化层在脱溶剂化过程中, 脱去所需要克服的能量比脱去EC 分子高, 即锂离子会保留在其溶剂化层中, 形成“离子接触溶剂化层”的界面电解液结构, 如图7(b)(左)所示. 与EC 不同的是, PC 分子由于其烷基基团的电荷密度略低于EC 子, 这点差异导致其与的结合能力较弱. 因此, 在PC 基电解液中, 锂离子脱溶剂化层过程中保留和PC 分子的能力相当. 相比较EC 体系, 后者在界面电解液中的含量较低(图7(b), 右). 更重要的是, EC 体系中含的“离子接触溶剂化层”得到电子发生还原反应时直接生成SEI 膜的主要成分LiF. 而PC 体系由于界面含量低, 因此产生的LiF 含量少, 不足以钝化石墨负极表面. 由此可见, 引起“PC-EC 在石墨负极界面差异”的根本原因是界面电解液中是否含有足够量的, 其形成的“离子接触溶剂化层”发生还原分解可产生具有钝化石墨负极SEI 膜的主要成分LiF.

图7 (a) 锂离子溶剂化层结构与石墨层间距的大小比例示意图; (b)引起“PC-EC 在石墨负极界面差异”的机理示意图[26]Fig. 7. (a) Schematic diagram of the scale between lithium ion solvation structure and graphite layer; (b) mechanism of "PC-EC disparity in the interface of graphite anode"[26].
锂离子电池在高电压及高温循环过程中, 含锂的过渡金属氧化物正极表面的过渡金属离子溶出加剧. 与锂离子相比较, 这些溶解的过渡金属离子电荷密度更高, 因此在电解液中形成比锂离子更加复杂的溶剂化层结构[27]. 根据离子的还原活性顺序可以推测, 这些过渡金属离子由于还原活性比锂离子高, 因此, 应该在锂离子嵌入石墨负极之前就被还原为金属, 并沉积在负极表面. 然而, 已报道的大部分实验却在石墨负极表面检测到+2 价的锰化合物[28,29]. 基于对溶剂化层中的组分影响溶剂/离子电化学稳定性的认识, 我们认为这种实验与理论结果的差异, 有可能是由于理论模型中缺乏考虑溶剂化层对离子还原活性的影响. 因此, 我们采用理论计算, 详细研究了Mn2+在电解液中形成的溶剂化层结构, 以及它对还原活性的影响[27]. 结果表明, Mn2+形成的溶剂化层得到电子时, 电子均主要分布在其溶剂化层的溶剂分子/阴离子上. 表明Mn2+催化其溶剂化层中的组分发生还原, 而自身以+2 价的化合物伴随溶剂/阴离子的还原产物沉积在负极表面. 值得一提的是, Mn2+离子在水溶剂中所形成的溶剂化层结构, 得到电子时, Mn2+离子优先被还原, 因此, 溶剂化层的组分对Mn2+离子还原活性的影响. 随后, 我们采用同样的计算方法研究了Co2+在碳酸酯基电解液中的还原和沉积行为[30]. 发现与Mn2+不同, Co2+既可以催化溶剂化层中的组分还原, 自身以+2 价化合物沉积在石墨负极, 也可以自身发生还原, 以Co 金属沉积在石墨负极. 而且后者更容易进行. X 射线光电子能谱结果证明石墨负极表面沉积的钴既有金属钴也有二价钴化合物.
综上所述可知, 电解液中的离子以及溶剂分子的还原活性、分解产物均受到其溶剂化层中的组分所影响. 这种影响可能会降低研究体系的还原稳定性和改变还原产物, 例如会降低锂离子溶剂化层的还原稳定性, 且形成LiF 的分解产物; 过渡金属Mn2+离子会催化降低其溶剂化层中的碳酸酯分子/发生还原分解. 因此, 在研究电解液的还原稳定性、分解成膜机理, 或者是设计和预测新型电解液的还原稳定性时, 必须考虑其溶剂化层的影响.
5 实验方法测试电解液电化学稳定性需要注意的问题
在实验研究中, 可以通过测试和比较电解液的循环伏安(C-V), 线性扫描(LSV), 充放电或微分容量 (dQ/dV) 曲线, 较为直观地判断电解液中某一组分/成膜添加剂的电化学稳定性. 如图8(a)中,与空白电解液体系(不含添加剂)相比较, 含苯基磷酸二甲酯(DMPP)添加剂的电解液在锰酸锂电极上的首次dQ/dV曲线中出现一个新的氧化峰[31].因此, 该氧化峰可以认为是DMPP 添加剂的氧化反应(其氧化分解是否成膜, 以及膜的性质, 还需要结合后续电化学性能说明). 图8(b)的LSV 曲线中, 与空白电解液体系相比较, 含N-烯丙基-N,N-双(三甲基甲硅烷基)胺(NNB)添加剂的电解液在Pt 电极上, 在4.2 V 左右出现一个氧化峰. 该峰可以归结于NNB 添加剂的氧化分解[32]. 与此同时, 还可以发现, 该电解液体系在后续高电位的氧化电流明显低于空白体系, 表明NNB 的氧化分解产物有效钝化Pt 电极表面, 进而保护本体电解液在高电位条件下不被氧化.
然而, 需要指出的是, 有一部分电解液组分/成膜添加剂的电化学分解反应速率较慢, 采用上述的电化学研究方法无法检测到其发生电化学分解的电流峰/电位平台. 这种情况下就需要结合其他的研究进一步验证其作用机理, 而不能简单地认为体系没有相应的电流峰/电位平台就是没有发生电化学反应. 例如, 二氟磷酸锂(LiPO2F2)是一种有效的成膜添加剂, 其构筑的界面膜不仅可以有效地抑制电解液在高电压条件下的氧化分解, 还可以显著提高离子在界面的扩散速率, 进而提高电池的倍率性能[33]. 虽然理论计算结果表明, LiPO2F2的氧化活性比碳酸酯基电解液的组分都高(即具有较低的计算氧化电位)(图8(c)), 理论上会优先于碳酸酯基电解液发生氧化分解. 但是, 其在Pt 电极上的LSV 曲线观测不到相应的氧化峰电流, 在三元正极上的C-V曲线也与空白电解液体系相似(图8(d)).因此, 难以判断LiPO2F2是否发生了氧化分解. 因此, 我们测试电解液中的19F 核磁共振谱在电化学测试前后的变化, 以监测LiPO2F2在电化学循环前后含量的变化, 证明LiPO2F2确实在循环后被消耗了(图8(e)). 更重要的是, LiPO2F2的“消耗反应”可以有效地抑制电解液在高电压三元正极材料表面的氧化分解(图8(f)). 因此, 结合上述的理论与实验结果, 我们提出LiPO2F2确实通过优先氧化分解, 在高电压正极表面形成了稳定的界面膜,是一种高效的成膜添加剂, 只是由于其在所研究电极表面的分解速率较慢, 因此难以检测到其相应的氧化峰电流.
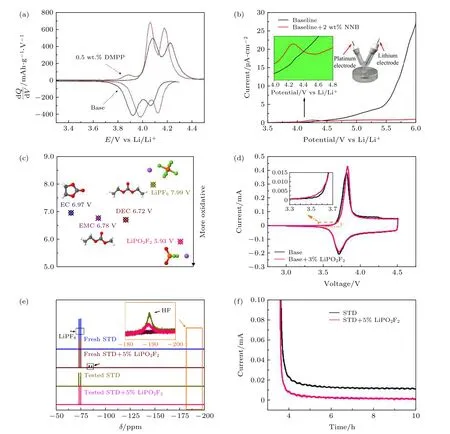
图8 (a) DMPP 添加剂在锰酸锂电极上的C-V 曲线; (b) NNB 添加剂在Pt 电极上的LSV 曲线; (c) LiPO2F2 与电解液其他组分的计算氧化电位; (d) LiPO2F2 添加剂在三元正极上的CV 曲线; (e) 电解液在经过电化学测试前后的19F 核磁共振谱; (f) 三元正极在不同电解液中的计时电流曲线[31—33]Fig. 8. (a) C-V curves of DMPP additive on LMO electrode; (b) LSV curves of NNB additive on Pt electrode; (c) calculated oxidation potential (V vs. Li/Li+) of EC, EMC, DEC, LiPF6 and LiPO2F2; (d) C-V curves of LNCM/Li cells with and without LiPO2F2;(e) 19 F NMR spectra of electrolytes before and after electrochemical test; (f) chronoamperometric responses of LNCM/Li cells with and without LiPO2F2[31—33].
因此, 电解液成膜添加剂在电化学测试过程中能否出现相应的电流峰/电压平台, 不仅与其电化学稳定性相关, 还受分解速率的影响. 在实验测试过程中没有出现相应电流峰/电压平台的添加剂,有可能也参与成膜反应, 需要结合多种表征方法研究其作用机理.
6 结 论
界面电解液的电化学稳定性是决定电极/电解液界面性质及电池性能的关键因素. 因此, 研究界面电解液的电化学稳定性及分解反应机理, 有助于解析界面性质及电池性能变化的原因. 更重要的是, 为下一代新型电解液体系的设计提供理论指导依据. 根据上述已有的研究结果可知, 在分析界面电解液电化学性质时, 必须考虑离子溶剂化层中各组分之间的影响, 特别是溶剂化层中的影响.界面电解液的分解机理和影响因素非常复杂, 至今仍有许多内容需要去进一步研究, 例如电极材料如何影响界面电解液的稳定性和分解机理; 界面的分解产物、结构与界面性质的关系; 成膜添加剂电化学反应速率与其形成的界面性质的关系等. 因此,本文最主要的目的在于抛砖引玉, 希望可以引起更多的科学家关注和重视电解液及其界面性质的研究.
猜你喜欢
电池(2022年4期)2022-11-07
汽车工程师(2021年12期)2022-01-18
汽车工程师(2021年12期)2022-01-18
汽车工程师(2021年12期)2022-01-18
电动工具(2020年2期)2020-04-22
储能科学与技术(2020年2期)2020-04-04
科学与技术(2019年13期)2019-04-19
中国校外教育(中旬)(2018年9期)2018-09-30
汽车电器(2014年5期)2014-02-28
储能科学与技术(2014年5期)2014-02-27
