高效液相色谱-串联质谱法测定动物源性食品中矮壮素残留
2020-12-25 02:41吴海智袁列江唐吉旺王淑霞
色谱 2020年2期
肖 泳,吴海智,袁列江,唐吉旺,王淑霞,王 秀,邓 航,吴 林
(湖南省产商品质量监督检验研究院,湖南 长沙 410007)
矮壮素(chlormequat chloride,CCC,分子结构式见图1)是一种季铵盐类植物生长调节剂,可用于小麦、水稻、棉花、烟草、玉米及西红柿等作物,能明显改善农作物品质,提高产量。然而,矮壮素可引起人和动物急性中毒,并对动物的繁殖能力有不良影响,美国国家职业安全和健康研究所(NIOSH)已将矮壮素列为疑似内分泌干扰物质。因此,食品中矮壮素的残留已成为影响食品安全的重要潜在因素,许多国家和地区对这类农药在食品中的残留量制定了严格的限量标准。国际食品法典委员会(CAC)制定了包括谷物及其加工制品、油料以及动物产品中矮壮素的限量标准;欧盟制定了包括谷物及其加工制品、油料、动物产品、蔬菜、水果、茶叶、调味品等10类产品中矮壮素的限量标准;日本制定了谷物及其加工制品、油料、动物产品、蔬菜、水果等9类产品中矮壮素的限量标准。2018年,美国环保署不同基质中对矮壮素最大残留限量进行了修订,包含大麦、燕麦、小麦、猪肉、牛肉、山羊肉、绵羊肉、猪肉副产品、牛肉副产品、山羊肉副产品、绵羊肉副产品、牛奶、禽肉、禽肉副产品、蛋;新版国家标准GB 2763-2019中对谷物、油料和油脂、蔬菜中矮壮素最大残留限量作了规定,且新增了哺乳动物肉类及内脏(海洋哺乳动物除外)、禽肉类、禽类内脏、蛋类、生乳的临时限量,但该标准中只有谷物、油料和油脂、蔬菜的测定方法。
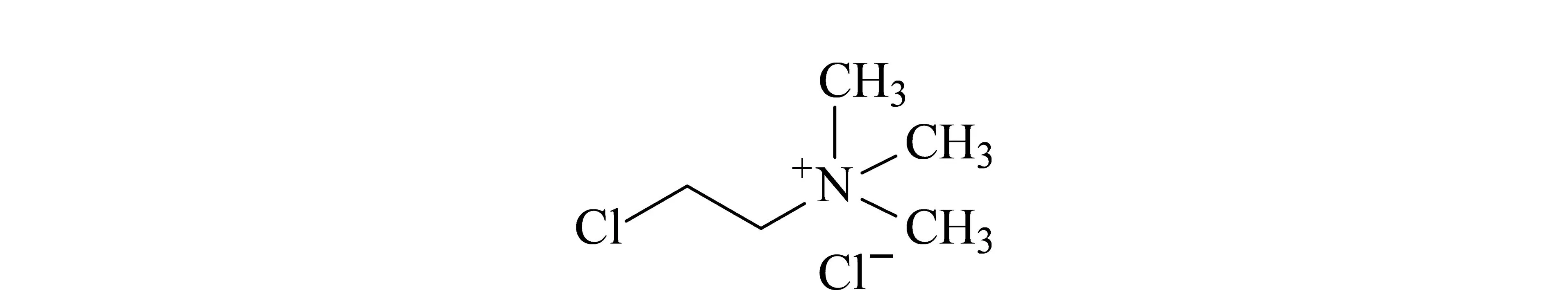
图1 矮壮素的分子结构式Fig.1 Molecular structure formula of chlormequat chloride (CCC)
目前,国内外关于食品中矮壮素的检测方法主要有气相色谱-质谱法[1]、液相色谱-串联质谱法[2-17]和离子色谱法[18]等。离子色谱法灵敏度没有气相色谱-质谱法和液相色谱-质谱法高,气相色谱-质谱法需要衍生,液相色谱-串联质谱法前处理相对简便且具有高特异性和高灵敏度。矮壮素常用的前处理方法有直接萃取法[3,6,7]、QuEChERS法[4,5,17]、固相萃取法(SPE)[11-16]等,常用的固相萃取柱有阳离子交换柱[13,14]、弱阳离子交换柱[11,12]、HLB固相萃取小柱[15,16]等,但现有文献方法大多针对蔬菜、水果、粮谷类样品,对动物源性食品中矮壮素检测的研究很少。本文以动物源性食品为研究对象,采用内标法定量,建立了高效液相色谱-串联质谱测定动物源性食品中矮壮素残留的分析方法,并研究了色谱柱、提取溶剂、净化方式、固相萃取柱等因素对矮壮素提取效率及净化效果的影响。
1 实验部分
1.1 仪器、试剂与材料
TSQ Quantum Ultra-AM高效液相色谱-串联质谱仪(美国Thermo公司);MV5多通道氮吹浓缩仪(莱伯泰科有限公司);DHS-220双模式自动均质仪(北京莱伯泰科有限公司);Allegra 64R高速冷冻离心机(美国Beckman Coulter公司);GM200刀式研磨仪(德国Retsch公司);Venusil MP C18(2)色谱柱(150 mm×2.1 mm,3 μm)、Agela Cleanert PCX固相萃取柱(60 mg/3 mL)(天津博纳艾杰尔科技有限公司)。
矮壮素(纯度99.0%)、矮壮素-D4(纯度99.3%)(德国Dr.Ehrenstorfer公司);乙腈、正己烷、甲醇、甲酸、乙酸(HPLC级,上海安谱公司);乙酸铵(HPLC级,美国ROE公司);ODS C18粉末(50 μm,天津博纳艾杰尔科技有限公司);无水硫酸镁、无水乙酸钠、氯化钠(分析纯,国药集团有限公司)。
称取适量矮壮素和矮壮素-D4标准品,分别用甲醇配制成1.00 g/L的标准储备液,于0~4 ℃冰箱中储存。根据需要将矮壮素标准储备液用乙腈稀释成适当浓度的标准工作液,将矮壮素-D4内标储备液用乙腈稀释成1.00 mg/L的内标工作液,于0~4 ℃冷藏储存,备用。
1.2 实验方法
1.2.1样品制备
猪、牛、羊、鸡等畜禽类样品取可食肌肉组织、内脏、蹄翅(去骨)约200 g,切碎后经刀式研磨仪充分搅碎混匀,装入洁净容器内,密封,于-18 ℃以下冷冻保存。蛋类取10枚,去壳后经刀式研磨仪充分混匀,装入洁净容器内,密封;牛奶样品取约200 g充分混匀,装入洁净容器内,密封,蛋类和牛奶样品置于0~4 ℃冷藏保存。
1.2.2样品前处理
称取5 g(精确至0.01 g)制备均匀的样品,置于50 mL具塞离心管中,准确加入50 μL CCC-D4内标溶液(1.00 mg/L),混匀静置20 min;加入15 mL含1%(v/v)乙酸的乙腈溶液,均质2 min后超声5 min,加入1~2 g氯化钠、2~3 g无水硫酸镁,均质2 min,用10 mL乙腈清洗均质头和超声棒体,清洗液并入样品离心管中,离心,收集提取液;残渣加入15 mL含1%(v/v)乙酸的乙腈溶液,均质提取2 min,并用10 mL乙腈清洗均质头,清洗液并入样品离心管中,离心,合并两次提取液并用乙腈定容至50 mL;取15 mL提取液,用10 mL乙腈饱和的正己烷脱脂两次,准确移取10 mL提取液,于40 ℃氮吹浓缩至干,加入3 mL水溶解样品,待进一步净化。
用3 mL甲醇和3 mL水活化PCX固相萃取柱,上样,用3 mL水和3 mL甲醇淋洗除杂,用5 mL甲醇(含100 mmol/L乙酸铵)溶液洗脱;洗脱液于40 ℃氮吹浓缩至干,用1 mL乙腈-0.1%(v/v)甲酸水溶液(1∶1,v/v)溶解残渣,过0.22 μm有机滤膜后,供HPLC-MS/MS测定。
1.2.3色谱和质谱条件
色谱柱:Venusil MP C18(2)柱(150 mm×2.1 mm,3 μm);柱温:40 ℃;流速:0.3 mL/min;流动相:A为0.1%(v/v)甲酸水溶液,B为乙腈。梯度洗脱程序:0~1.00 min,10%B;1.00~3.50 min,10%B~95%B;3.50~4.00 min,95%B;4.00~4.01 min,95%B~10%B;4.01~5.00 min,10%B。进样体积:10 μL。
离子源:电喷雾电离(ESI)源;扫描模式:正离子扫描;扫描方式:多反应监测(MRM);毛细管温度:350 ℃;蒸发温度:300 ℃;鞘气流量:9.45 L/min;辅助气流量:12.1 L/min;喷雾电压:3 500 V;其他质谱条件见表1。
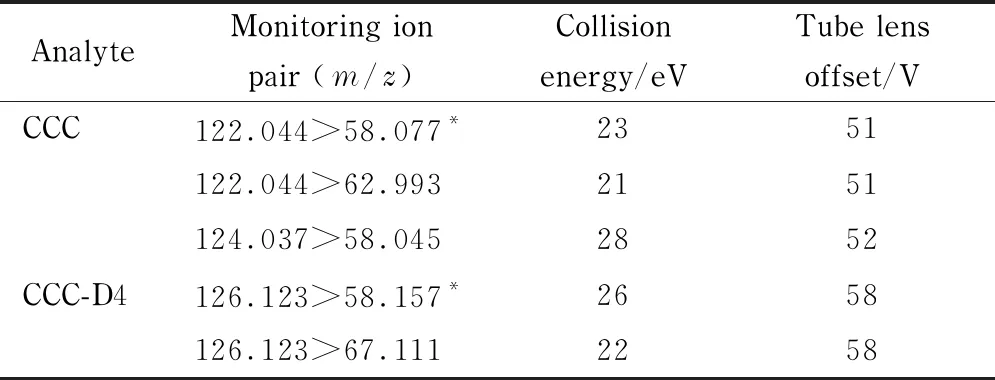
表1 CCC和CCC-D4的质谱参数Table 1 MS parameters of CCC and CCC-D4
* Quantitative ion.
2 结果与讨论
2.1 色谱条件的选择
2.1.1色谱柱的选择
矮壮素常采用的色谱柱有C18柱[3,7,14,15,17]、HILIC柱[6,8,10-12]等,本方法以加标鸡蛋样品为研究对象,在流动相均采用0.1%(v/v)甲酸水溶液(含10 mmol/L乙酸铵)和乙腈的条件下,比较了Venusil MP C18(2)(150 mm×2.1 mm,3 μm)、Venusil HILIC(150 mm×2.1 mm,3 μm)、Thermo Hypersil Gold(150 mm×2.1 mm,3 μm)3种色谱柱对矮壮素的保留能力和出峰效果(见图2)。Venusil HILIC柱分离时,采用0.1%(v/v)甲酸水溶液(含10 mmol/L乙酸铵)-乙腈(40∶60,v/v)等度洗脱;采用Venusil MP C18(2)柱和Thermo Hypersil Gold柱分离时,采用1.2.3节的梯度洗脱程序。结果表明,3种色谱柱的出峰时间差别不大,但是采用Venusil MP C18(2)色谱柱时,矮壮素的峰形比采用另外两种色谱柱时更好、更对称。因此,选择Venusil MP C18(2)色谱柱作为矮壮素的分离色谱柱。
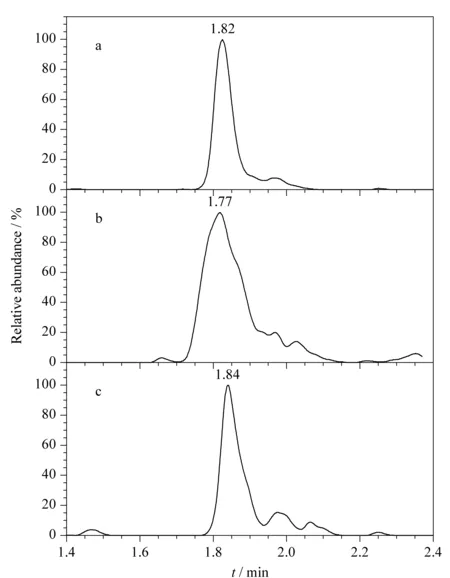
图2 采用不同色谱柱时加标鸡蛋样品中矮壮素(0.500 μg/kg)的色谱图Fig.2 Chromatograms of CCC (0.500 μg/kg)spiked in egg samples using the different chromatographic columns a.column:Venusil MP C18(2)(150 mm×2.1 mm,3μm);mobile phase:(A)0.1% (v/v)formic acid aqueous solution (containing 10 mmol/L ammonium acetate)and (B)acetonitrile;gradient elution program:0-1.00 min,10%B;1.00-3.50 min,10%B-95%B;3.50-4.00 min,95%B;4.00-4.01 min,95%B-10%B;4.01-5.00 min,10%B.b.column:Venusil HILIC (150 mm×2.1 mm,3μm);mobile phase:0.1% (v/v)formic acid aqueous solution (containing 10 mmol/L ammonium acetate)-acetonitrile (40∶60,v/v);isocratic elution.c.column:Thermo Hypersil Gold (150 mm×2.1 mm,3 μm).Mobile phases and gradient elution program were the same as that in Fig.2a.
2.1.2流动相的选择
实验分别考察了流动相为水-乙腈、0.1%(v/v)甲酸水溶液-乙腈、0.1%(v/v)甲酸水溶液(10 mmol/L乙酸铵溶液)-乙腈时矮壮素的分离效果。实验发现,含有0.1%(v/v)甲酸水溶液的流动相能有效提高矮壮素的离子化效率,增强检测灵敏度,而乙酸铵的加入对样品中矮壮素的分离并没有明显的影响。因此,实验选择0.1%(v/v)甲酸水溶液-乙腈作为流动相。
2.2 提取溶剂的选择
矮壮素具有强极性,易溶于水、甲醇和乙腈等溶剂,常采用甲醇-水、甲醇-水-乙酸溶液、乙腈等溶剂进行提取。李春梅等[12]比较了纯水和不同体积分数的甲醇水溶液、乙腈水溶液对矮壮素回收率的影响,最终得出用乙腈作为小鼠肌肉中矮壮素的提取溶剂效果最佳。考虑到动物源性食品中蛋白质含量均较高,本研究用含1%(v/v)乙酸的乙腈溶液作为提取溶剂,以进一步沉淀样品中的蛋白质;考虑到动物源性食品中脂肪含量均较高,用正己烷对提取液进行脱脂处理后,发现提取液明显变得澄清,且不会影响矮壮素的回收率。因此,实验选择含1%(v/v)乙酸的乙腈溶液作为提取溶剂,并用正己烷对提取液进行脱脂处理。
2.3 净化方式的选择
矮壮素常用的前处理方法有直接萃取法、QuEChERS法和SPE法等。实验尝试采用含1%(v/v)乙酸的乙腈溶液提取、浓缩后直接过膜上机测定,结果发现,由于动物源性样品中脂肪和蛋白质含量较高,提取液浓缩复溶后不利于过膜,且基质抑制作用明显,杂质峰干扰大,不利于定量;采用含1%(v/v)乙酸的乙腈溶液提取,并加入QuEChERS盐包(含无水硫酸镁、无水乙酸钠),提取液再经QuEChERS净化管(含C18粉末和无水硫酸镁)净化,浓缩后过膜上机测定,结果发现,该方法对动物源性样品的净化效果不是特别理想,目标峰尾部有小的干扰峰,方法灵敏度较SPE法低,QuEChERS法采用内标法定量时,方法回收率能达到80.0%以上,可适用于大批量样品的筛查;采用SPE法能有效去除样品中的杂质,方法灵敏度高,回收率好。因此,实验选择SPE法作为前处理方法。
矮壮素常用的固相萃取柱有阳离子交换柱、弱阳离子交换柱、HLB固相萃取小柱等。本研究比较了Supelclean LC-WCX固相萃取柱(500 mg/3 mL)、CNW Poly-Sery HLB固相萃取柱(200 mg/6 mL)、Agela Cleanert PCX固相萃取柱(60 mg/3 mL)的净化效果和回收率。采用WCX柱作为净化柱时,矮壮素的回收率为85.2%,与李春梅等[12]的研究结果一致。本方法按内标法计算时,回收率接近100%,但是该柱在净化肝肾样品时,色谱峰尾部有小的干扰峰。杨涛等[16]选择HLB柱作为净化柱,用5%(v/v)甲醇水溶液淋洗和甲醇洗脱后方法的回收率为82.5%~104%,而本研究发现,用5%(v/v)甲醇水溶液淋洗时矮壮素和内标均有不同程度的损失,最终回收率不到40.0%,这与杨涛等[16]的研究结果不一致;本研究采用曹慧等[15]的方法对动物源性食品进行净化,选择HLB柱作为净化柱,并未淋洗而是直接用5%(v/v)氨水甲醇溶液洗脱,发现目标峰尾部有干扰峰,不利于定量。李娜等[14]选择PCX柱作为净化柱,用水、50%(v/v)甲醇水溶液、甲醇淋洗,50%(v/v)甲醇水溶液(100 mmol/L乙酸铵)洗脱,方法的回收率为90.5%~98.5%,本研究发现对于动物源性食品采用李娜等[14]的净化方法能有效去除样品中的杂质,方法回收率可达90%以上。因此,实验最终选择PCX固相萃取小柱进行净化。
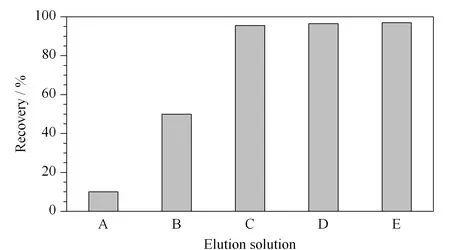
图3 不同洗脱溶液对矮壮素回收率的影响Fig.3 Effects of different elution solutions on the recoveries of CCC A.methanol (containing 50 mmol/L ammonium acetate);B.methanol (containing 75 mmol/L ammonium acetate);C.methanol (containing 100 mmol/L ammonium acetate);D.50% (v/v)methanol aqueous solution (containing 100 mmol/L ammonium acetate);E.80% (v/v)methanol aqueous solution (containing 100 mmol/L ammonium acetate).
矮壮素在PCX小柱中保留能力较强,用甲醇无法直接洗脱下来,需加入一定量的乙酸铵。研究采用PCX柱作为净化柱,以水、甲醇为淋洗液,以含不同浓度乙酸铵的甲醇溶液为洗脱溶液进行净化,方法的回收率如图3所示,随着洗脱溶液中乙酸铵浓度的增加,矮壮素的回收率从10.0%升至95.5%;实验还比较了洗脱溶液中的含水量对矮壮素回收率的影响,研究发现,洗脱溶液含水量对矮壮素的回收率无影响,考虑到洗脱溶液需再浓缩,因此选择甲醇(含100 mmol/L乙酸铵)作为洗脱溶液。
2.4 线性范围与定量限
按1.2节方法制备猪肉、牛肉、羊肉、鸡肉、鸡蛋、猪肾、牛肝、羊肾、鸡肝、牛奶样品的空白基质溶液,配制成质量浓度分别为0.200、0.500、1.00、5.00、10.0、50.0、100、200、500 μg/L的基质匹配标准溶液(含内标10.0 μg/L),对矮壮素和矮壮素-D4定量离子的峰面积之比(y)与矮壮素的质量浓度(x,μg/L)进行线性回归,绘制标准曲线,其线性方程及相关系数(r2)见表2。
结果表明:矮壮素在0.200~500 μg/L范围内线性关系良好,其线性相关系数均不低于0.999 3,优于李春梅等[12]方法的线性范围和相关系数。
在阴性样品中添加矮壮素,以10倍信噪比(S/N≥)计算方法的定量限(LOQ),结果如表2所示,其LOQ为0.500 μg/kg,与李春梅等[12]方法相比具有更高的灵敏度。
2.5 准确度与精密度
通过对阴性样品进行加标回收,考察方法的准确度和精密度。按照1.2节提取和净化步骤,在猪肉、牛肉、羊肉、鸡肉、鸡蛋、猪肾、牛肝、羊肾、鸡肝和牛奶10种基质样品中分别添加定量的矮壮素标准溶液进行加标回收试验,每个加标水平做6个平行(见表2),加标水平为0.500 μg/kg时的色谱图见图4。
结果显示,在3个加标水平下,猪肉、牛肉、羊肉、鸡肉、鸡蛋、猪肾、牛肝、羊肾、鸡肝和牛奶中矮壮素的平均回收率为93.4%~101%,相对标准偏差为2.3%~8.0%。说明方法的准确度高,通用性好,且方法的回收率明显优于李春梅等[12]方法的回收率。
2.6 实际样品的测定
运用本文建立的方法,对市售的猪肉、牛肉、羊肉、猪肝、猪肾、牛肝、羊肾、鸡肉、鸭肉、鸡胗、鸡肝、鸡翅、鸭肠、鸡蛋、鸭蛋、牛奶共43份样品进行矮壮素的残留检测。其中,5份不同厂家生产的鸡蛋样品中有1份检出矮壮素,含量为1.31 μg/kg;6份不同厂家生产的牛奶样品中有4份检出矮壮素,含量分别为3.32、4.02、6.63和2.01 μg/kg,其余样品未检出矮壮素,所检出样品中矮壮素的含量均低于国家标准对其规定的最大残留限量。
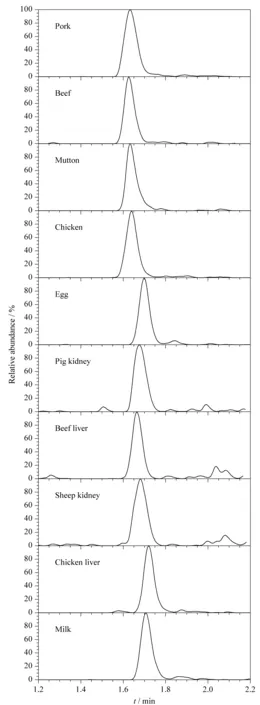
图4 不同加标样品中矮壮素(0.500 μg/kg)的色谱图Fig.4 Chromatograms of CCC (0.500 μg/kg)spiked in the different samples
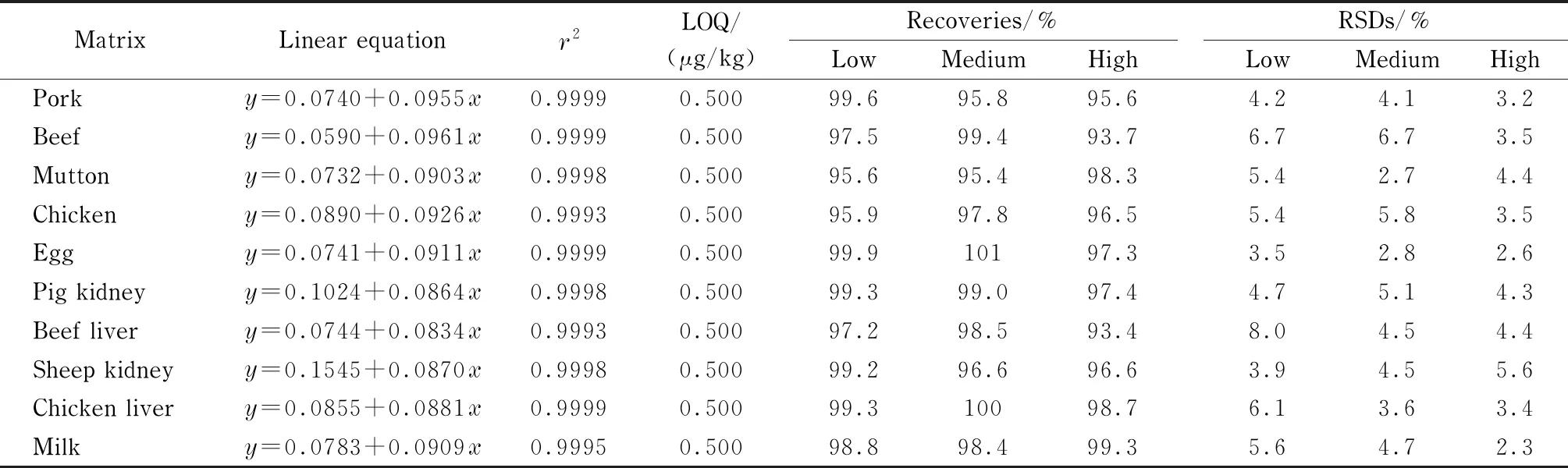
表2 不同基质中矮壮素的线性方程、相关系数、定量限、加标回收率和精密度(n=6)Table 2 Linear equations,correlation coefficients (r2),LOQs,spiked recoveries and precisions of CCC in the different matrices (n=6)
Low,medium,high spiked levels:0.500,10.0,200 μg/kg for pork,beef,mutton;0.500,5.00,40.0 μg/kg for chicken;0.500,10.0,100 μg/kg for egg,chicken live;0.500,20.0,500 μg/kg for pig kidney,beef liver,sheep kidney,milk;y:peak area ratio of CCC to CCC-D4;x:mass concentration,μg/L.
3 结论
本文建立了高效液相色谱-串联质谱测定动物源性食品中矮壮素残留的分析方法,方法操作简单,净化效果好,定性准确,灵敏度高,线性关系、准确度和精密度均满足方法学指标,方法定量限完全满足各国限量标准要求,可为动物源性食品中矮壮素的残留检测提供技术手段。
猜你喜欢
煤化工(2022年3期)2022-07-08
中国饲料(2022年5期)2022-04-26
色谱(2021年7期)2021-06-07
陕西农业科学(2020年11期)2020-12-25
中国中医急症(2019年10期)2019-05-21
农家科技中旬版(2019年2期)2019-05-08
山东工业技术(2016年10期)2016-09-06
中国资源综合利用(2016年10期)2016-01-22
中国医疗美容(2015年1期)2015-07-12
癌变·畸变·突变(2015年4期)2015-02-27