
(n = 0—4)团簇的电子结构、成键性质及稳定性*
2021-01-28 08:13张超江许洪光徐西玲郑卫军
物理学报 2021年2期
张超江 许洪光 徐西玲† 郑卫军‡
1) (中国科学院化学研究所, 北京分子科学研究中心, 分子反应动力学国家重点实验室, 北京 100190)
2) (中国科学院大学, 北京 100049)
3) (北京怀柔综合性国家科学中心, 物质科学实验室, 北京 101400)
(2020 年8 月17 日收到; 2020 年9 月1 日收到修改稿)
1 引 言
过渡金属碳化物(transition metal carbide)是一类具有高熔点、高硬度、高热稳定性以及类金属性质的物质, 广泛应用于机械切割、高温部件以及核反应堆等领域, 在开发新型超高温陶瓷材料、二维材料、电子材料、能源材料以及催化材料等方面具有重要意义[1−7].近年来, 人们对过渡金属碳化物相关团簇已经进行了大量的研究[8−26].Guo等[9,10]在钛/碳团簇质谱中发现了具有特殊稳定性的团簇, 并推测其结构是一个具有高对称性(Th)的十二面体金属碳笼(Metcar).Reddy 等[11]采用密度泛函(density functional theory, DFT)方法研究了Ti8C12的能量、电子结构以及磁性等性质, 认为其稳定性主要归功于碳-碳以及钛-碳之间类似共价键的作用力, 钛原子的存在使团簇具有一定磁性.Wang 研究组采用光电子能谱技术对第四周期过渡金属掺杂碳团簇进行了系统的研究.他们发现(x = 2—5)团簇呈环状结构[22], 而(n = 2—8)团簇中则是链状和环状结构共存[23], 对于后过渡金属如Fe/Cu/Au 掺杂的碳团簇更倾向于形成金属原子位于碳链末端的线性结构[24−26].Xu 等[27]结合光电子能谱和理论计算发现(n = 3—10)团簇的电子结合能呈现明显的奇偶性, 结构为链状和环状竞争共存.Redondo与其合作者[28−37]对第四周期过渡金属掺杂碳团簇(M = Sc, Ti, V, Fe, Co, Zn, n = 1—8)进行了理论研究, 发现团簇结构与过渡金属3d 层电子数量和碳原子数目相关.Zheng 研究组[38−40]对团簇的研究发现团簇呈立方体结构, 随着碳含量的增加,团簇稳定性逐渐增加.Wang 和Cheng[21]及Wang 等[41]对团簇的研究则说明钛/碳团簇负离子更倾向于在立方晶格的基础上生长.第五周期前过渡金属碳化物以及MoC 等团簇已有报道[42−51].Castleman 研究组[50]采用负离子光电子能谱结合密度泛函对(n = 4—9)团簇进行了研究, 并认为这些团簇是三维结构、平面结构以及线性结构共存.相较于铌—碳键,(n =4—9)团簇更倾向于形成铌-铌键.第五周期的后过渡金属碳化物团簇如PtnC–, Au2C2和Au(C≡C)nAu (n = 1—3)的研究亦有报道[52−56].Harding 等[53]采用振动光谱结合密度泛函理论对PtnC+(n =3—5)团簇结构进行了表征, 结果显示Pt3C+团簇是一个具有显著稳定性的平面三配位碳结构.León 等[55]采用高分辨光电子成像技术结合理论计算对Au(C≡C)nAu–/0(n = 1—3)团簇的研究表明,为线性类乙炔结构,团簇负离子为低对称链状结构:而中性Au2C4和Au2C6团簇则是高对称聚乙炔结构.Lu[57,58]报道了团簇的理论研究, 认为当n ≥ 4 时, 除了中性Pt5C2团簇外,其余团簇中碳-碳键均断裂.
钽/碳团簇目前也有一些研究.Gregory 研究组[59−62]采用光致电离效率谱对中性TamCn团簇的研究发现: 含钽较多的团簇中不含C2单元, 这与之前中性TamCn团簇的红外振动谱的结果一致.Aravind 等[63]通过分析TaC–负离子的光电子能谱, 确定TaC 团簇的电子亲和能(electron affinity,EA)为(1.928 ± 0.056) eV.He 研究组[64−66]采用质谱与光电子能谱结合高精度量子化学计算发现以及负离子团簇可以活化小分子N2和CH4.Chernyy 等[67]分析了中性Ta5C3团簇的红外光谱, 认为其第一电子激发态位于458 cm–1处.碳化钽(TaC)因具有超高熔点(4153.15 K)以及较高的转换温度(Tc= 10.35 K), 在超高温陶瓷以及超导材料方面具有潜在应用[68−72].但是, 有关钽/碳团簇电子结构的研究依然匮乏, 钽/碳团簇的生长机制以及团簇中各原子间的成键性质仍需更加深入的研究.本文采用负离子光电子能谱结合密度泛函方法对团簇进行了研究, 揭示了钽/碳团簇的电子结构、成键性质以及稳定性.
2 研究方法
2.1 实验方法
实验部分是在课题组自行搭建的直线式飞行时间质谱-磁瓶式光电子能谱仪上完成的, 该装置主要由激光溅射团簇源、飞行时间质谱仪以及磁瓶式光电子能谱仪组成[73].实验时钽/碳样品(直径为13 mm, 钽/碳摩尔比为1∶1)被放置于可以二维移动的样品槽中, 固体纳秒Nd:YAG 激光器(Continuum Surelite II-10)通过倍频产生的溅射激光(532 nm)经光学透镜聚焦后轰击样品表面产生等离子体.同时, 脉冲阀(general valve series 9)喷出高纯氦气(压力约为4 atm)与等离子体碰撞、冷却形成钽/碳团簇.钽/碳团簇经Skimmer 准直后进入加速区.钽/碳团簇负离子被加速后经飞行时间质谱仪分析产生钽/碳团簇负离子的质谱.(n = 0—4)团簇负离子经质量门选质以及减速器减速后进入脱附区与脱附激光(532 和266 nm)相互作用而脱附电子, 产生的光电子在磁场的作用下进入磁瓶式光电子能谱仪, 经光电子能谱仪分析产生特定团簇负离子的光电子能谱.我们使用相似条件下Bi–和Pb–离子的光电子能谱对(n =0—4)团簇负离子的光电子能谱进行标定.本装置中磁瓶式光电子能谱仪的分辨率在光电子动能为1 eV 处约为40 meV.
2.2 计算方法
3 结果与讨论
3.1 实验结果
图1 是在不同脱附激光能量(532 和266 nm)条件下获得的(n = 0—4)团簇负离子的光电子能谱.光电子能谱中第一个谱峰最高点所对应的电子结合能(electron binding energy, EBE)为团簇负离子的垂直脱附能(vertical detachment energy,VDE).团簇的绝热脱附能则是通过沿着光电子能谱的第一个谱峰的上升沿画一条重合的直线, 该直线与谱图基线相交处的电子结合能加上仪器分辨率获得.实验所得(n = 0—4)团簇负离子的垂直脱附能和绝热脱附能列于表1.
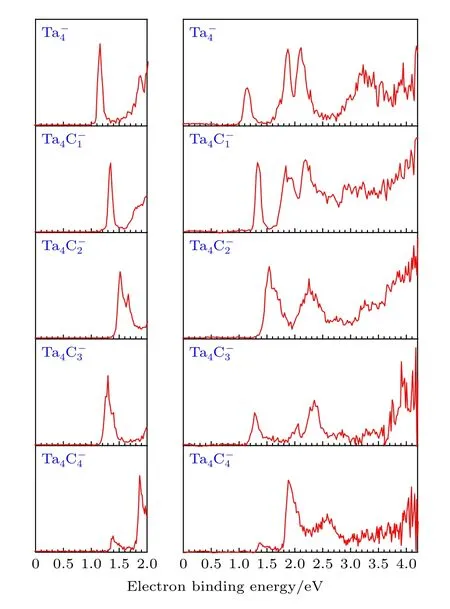
图1 在532 和266 nm 条件下采集的 (n =0—4)团簇负离子的光电子能谱Fig.1.Photoelectron spectra of (n = 0–4) cluster anions recorded with 532 (left) and 266 nm (right) photons.
表1 (n = 0—4)团簇负离子的低能量异构体的相对能量(ΔE), 理论VDEs/ADEs 以及实验VDEs/ADEsTable 1.Relativeenergies (ΔE), theoretical VDEs and ADEs of the low-lying isomers for (n = 0–4) cluster anions,as well as the experimental VDEs and ADEs estimated from their photoelectron spectra.
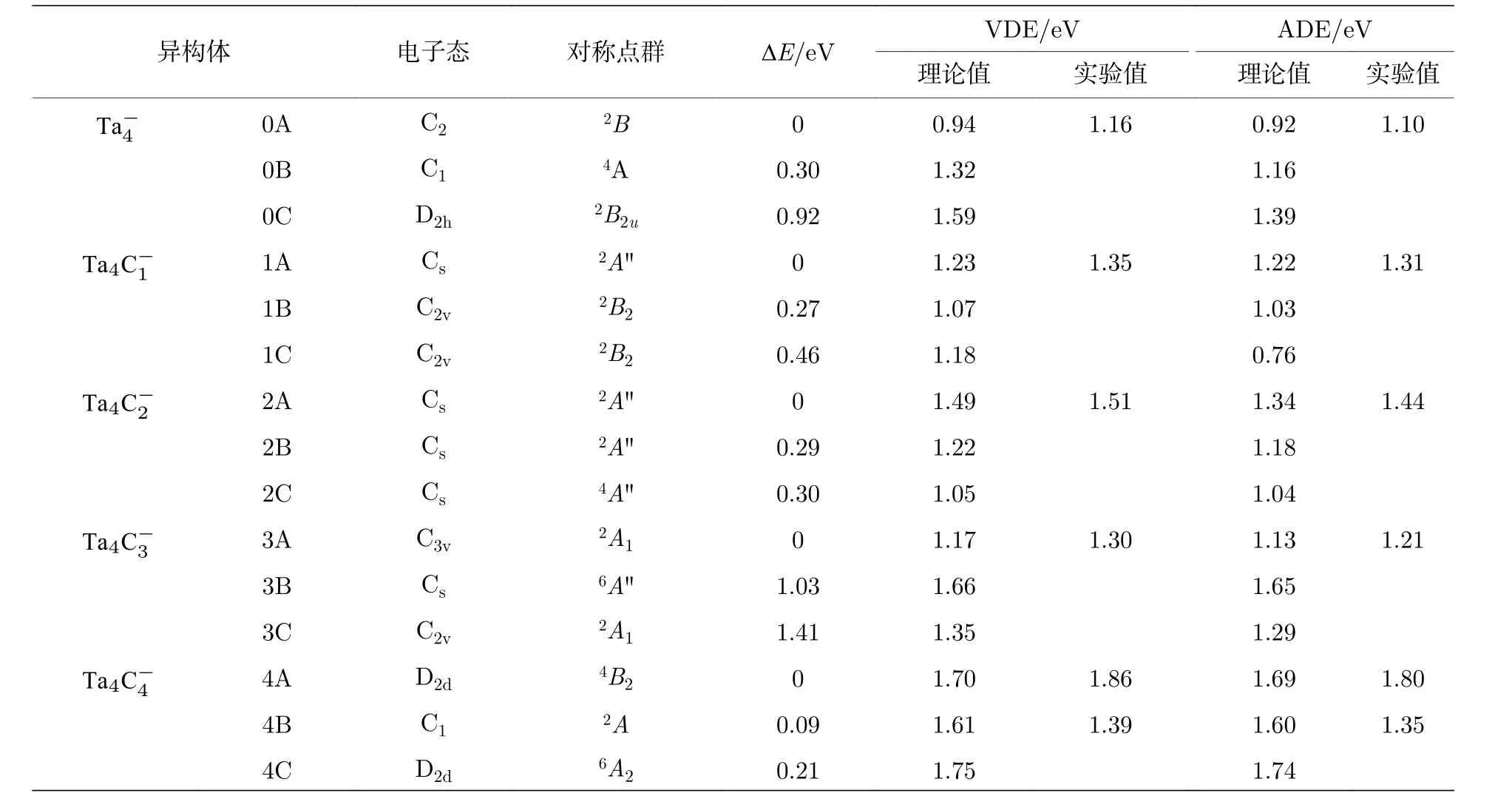
表1 (n = 0—4)团簇负离子的低能量异构体的相对能量(ΔE), 理论VDEs/ADEs 以及实验VDEs/ADEsTable 1.Relativeenergies (ΔE), theoretical VDEs and ADEs of the low-lying isomers for (n = 0–4) cluster anions,as well as the experimental VDEs and ADEs estimated from their photoelectron spectra.
异构体 电子态 对称点群 ΔE/eV VDE/eV ADE/eV理论值 实验值 理论值 实验值Ta-0A C2 2B 0 0.94 1.16 0.92 1.10 0B C1 4A 0.30 1.32 1.16 0C D2h 2B2u 0.92 1.59 1.39 Ta4C-4 1A Cs 2A"" 0 1.23 1.35 1.22 1.31 1B C2v 2B2 0.27 1.07 1.03 1C C2v 2B2 0.46 1.18 0.76 Ta4C-1 2A Cs 2A"" 0 1.49 1.51 1.34 1.44 2B Cs 2A"" 0.29 1.22 1.18 2C Cs 4A"" 0.30 1.05 1.04 Ta4C-2 3A C3v 2A1 0 1.17 1.30 1.13 1.21 3B Cs 6A"" 1.03 1.66 1.65 3C C2v 2A1 1.41 1.35 1.29 Ta4C-3 44A D2d 4B2 0 1.70 1.86 1.69 1.80 4B C1 2A 0.09 1.61 1.39 1.60 1.35 4C D2d 6A2 0.21 1.75 1.74
从图1 可以看出, 532 nm 条件下获得的光电子能谱具有较好的分辨率, 而266 nm 条件下的光电子能谱则包含了团簇在高电子结合能区域的电子结构信息.在团簇负离子的532 nm 光电子能谱上有两个中心位于1.16 和1.88 eV 的窄峰, 根据第一个谱峰确定了团簇负离子的实验VDE 和ADE 分别为(1.16 ± 0.08) eV 和(1.10 ±0.08) eV.除了532 nm 光电子能谱中的两个信号峰外, 在其266 nm 光电子能谱中还观察到两个信号较强的谱峰, 它们的电子结合能分别为2.11 eV和3.23 eV.在团簇负离子的532 nm 光电子能谱中含有一个窄峰以及一个肩峰, 其电子结合能分别为1.35 和1.84 eV.根据第一个谱峰确定团簇负离子的VDE 和ADE 分别为(1.35 ±0.08) eV 和(1.31 ± 0.08) eV.在团簇负离子的266 nm 光电子能谱中还可以观察到位于1.94 和2.19 eV 处的两个较宽的谱峰和一个中心位置位于3.09 eV 的谱带.在团簇负离子的532 nm 光电子能谱中可以观测到两个相邻的尖峰, 它们的电子结合能分别为1.51 和1.66 eV.团簇负离子的VDE 和ADE 分别为(1.51 ±0.08)和(1.44 ± 0.08) eV.532 nm 中的两个峰在其266 nm 光电子能谱中由于分辨率较低而无法分辨, 在266 nm 谱图中可以看到两个中心位于2.25 和3.25 eV 处较宽的特征峰.
3.2 理论结果

图2 (n = 0—4)团簇负离子的低能量异构体.相对能量是在PBEPBE/aug-cc-pVTZ/C/aug-cc-pVTZ-PP/Ta水平获得.其中红色球代表碳原子, 青色球代表钽原子Fig.2.Low-lying isomers of (n = 0–4) cluster anions.The relative energies are calculated at the PBEPBE/aug-ccpVTZ/C/aug-cc-pVTZ-PP/Ta level.Cyan and red balls stand for the tantalum and carbon atoms, respectively.
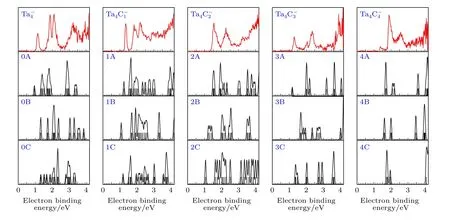
图3 (n = 0—4)团簇负离子的模拟光电子能谱(DOS)与实验光电子能谱对比, 竖线表示理论计算所对应的分子能级Fig.3.Comparisons of the experimental photoelectron spectra of (n = 0–4) with their simulated density of states (DOS)spectra.The vertical lines are the theoretically simulated spectral lines.

图4 中性Ta4Cn (n = 0—4)团簇的低能量异构体Fig.4.Low-lying isomers of neutral Ta4Cn (n = 0–4) clusters..
3.3 讨 论
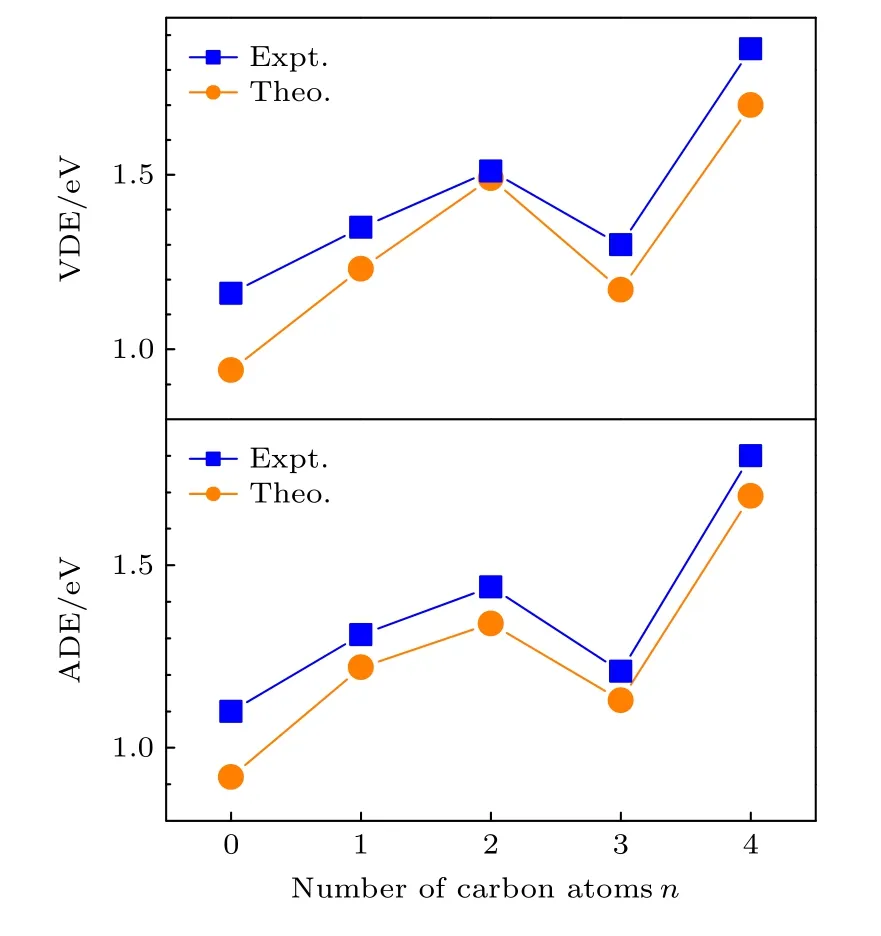
图5 (n = 0—4)团簇负离子的实验VDE/ADE和理论VDE/ADE 随碳原子增加的变化趋势Fig.5.Experimental and theoretical VDEs and ADEs of(n = 0–4) versus the number of carbon atoms.
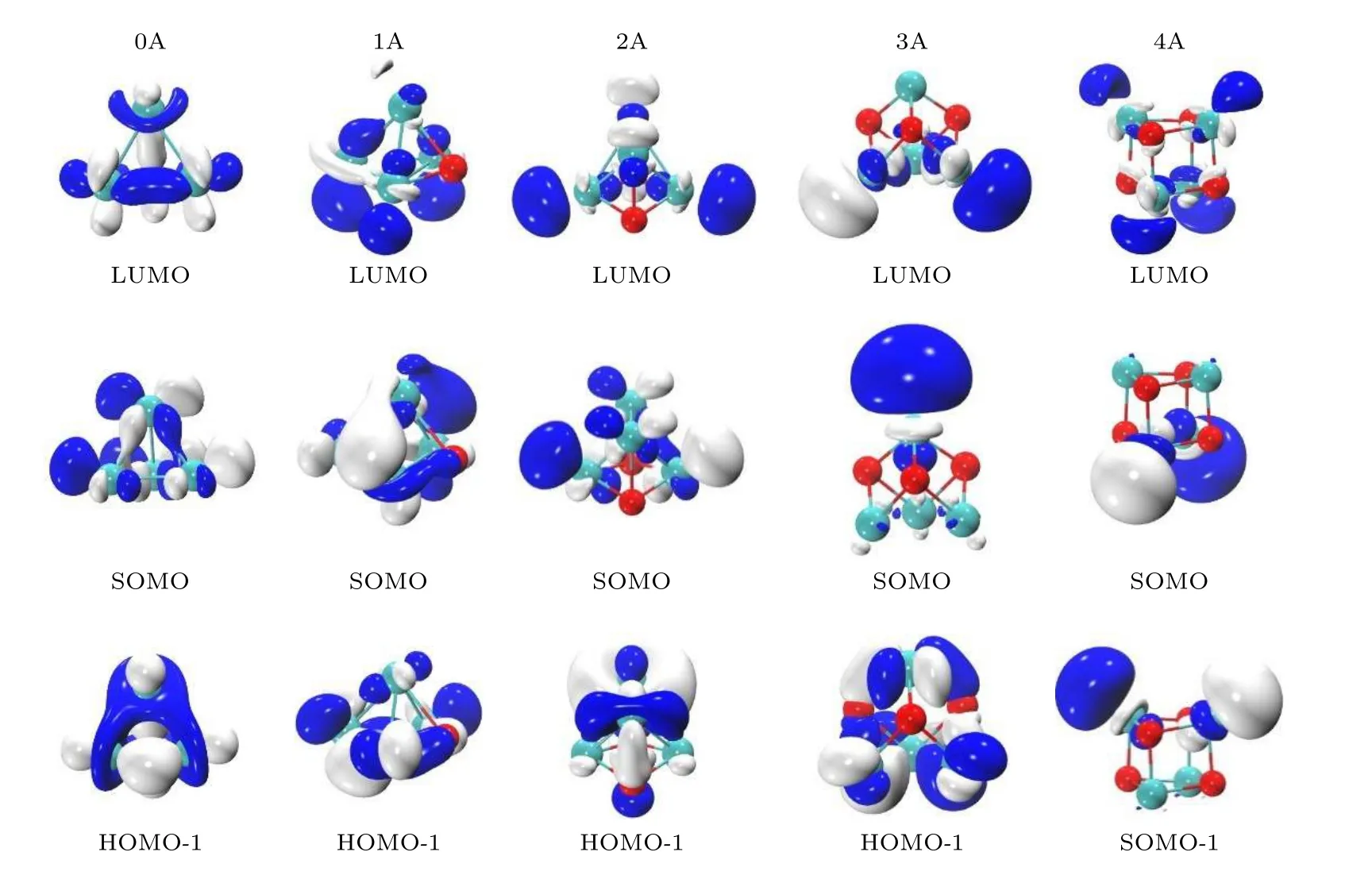
图6 (n = 0—4)团簇负离子的部分分子轨道示意图Fig.6.Diagrams of the selected molecular orbitals of (n = 0–4) cluster anions.
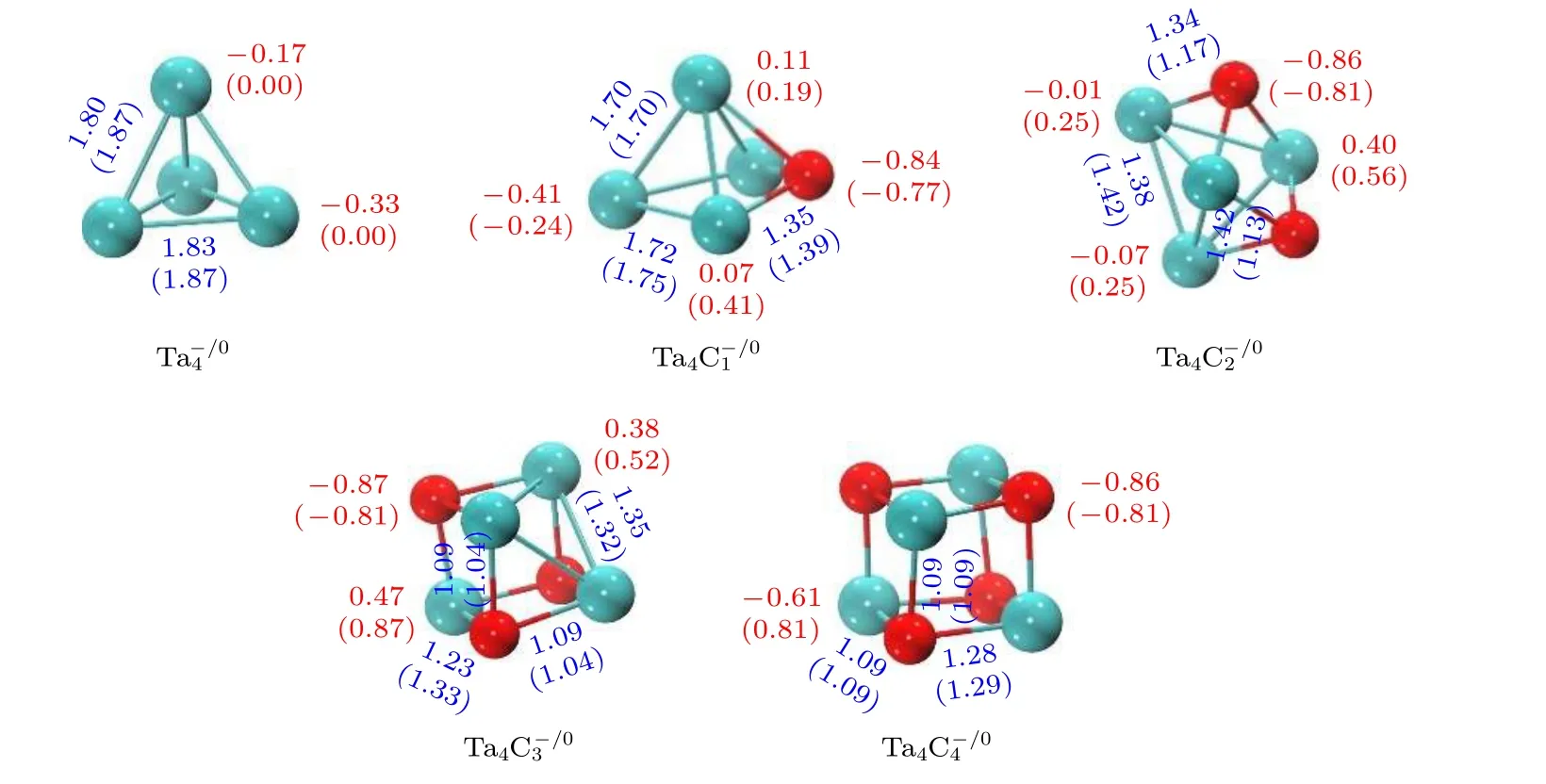
图7 (n = 0—4)团簇的NPA 电荷(Q,|e|, 红色数值)和Wiberg 键级(紫色数值), 括号中为中性团簇相对应数值Fig.7.NPA charges (Q, in|e|, red values) and Wiberg bond indices (WBIs, purple values) of the most stable structures of(n = 0–4) clusters.The values in parentheses are from the neutral clusters.
为了确认中性Ta4Cn(n = 0—4)团簇的结构,在PBE/aug-cc-pVTZ/C/aug-cc-pVTZ-PP/Ta 水平上对(n = 0—4)团簇正离子进行了优化,并获得中性团簇的电离能(ionization potentials,IPs).我们得到的中性Ta4Cn(n = 0—4)团簇的理论电离能分别为5.72, 5.75, 5.52, 5.64 和5.15 eV,与文献[61]中Ta4Cn(n = 0—4)团簇的实验电离能(5.83, 5.80, 5.55, 5.79 和5.15 eV)相符, 说明我们得到的中性团簇结构是合理的.中性Ta4Cn(n= 0—4)团簇最高占据分子轨道(highest occupied molecular orbital, HOMO)和最低未占分子轨道(lowest unoccupied molecular orbital, LUMO)能级差(HOMO-LUMO 能隙)分别为0.98, 0.72, 0.11,0.95 和0.03 eV.可以看到Ta4C2和Ta4C4团簇的HOMO-LUMO 能隙较小, 说明在团簇负离子的光电子能谱中第一个和第二个谱峰差距较小, 这与实验所得结果相吻合.
为了研究团簇的稳定性随碳原子增加的演变, 结合文献[62,83]中(n = 5—11)团簇的几何结构, 计算了和0—4)团簇的单原子结合能(Eb).计算方法如下:
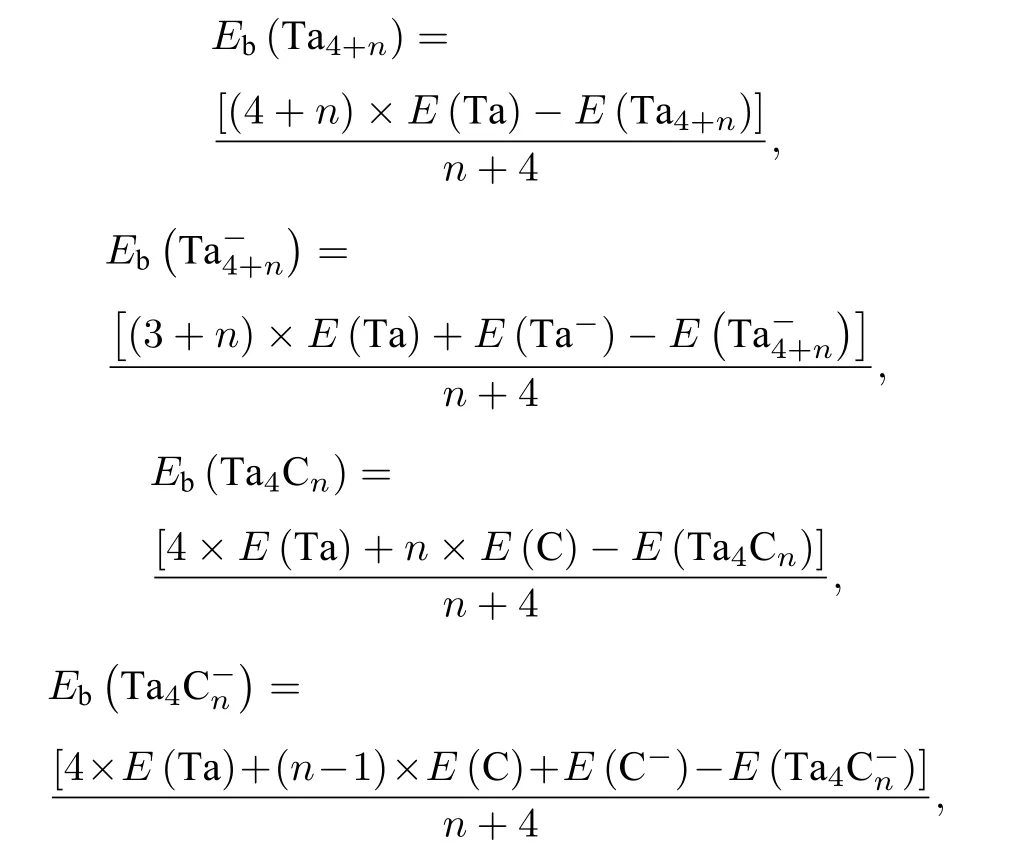
其中E 对应团簇或原子的能量, 所得结果如图8以及表2 所列.由图8 和表2 可以看出, 随着原子数目的增加,(n =0—4)团簇的Eb逐渐增加.这说明随着原子数目的增加, 团簇解离成单个原子所需能量逐渐增加.同时, 将团簇的Eb与纯金属团簇的Eb进行比较, 发现团簇的Eb远高于相应团簇的Eb, 中性Ta4C4团簇的单原子结合能高达7.13 eV, 而中性Ta8团簇的单原子结合能仅为5.37 eV.这说明用碳原子取代钽原子, 使得团簇解离成单个原子所需能量逐渐增加, 钽-碳共价键的形成有利于提高材料的熔点.这也印证了碳化钽的熔点(4153.15 K)远高于钽金属的熔点(3290.15 K)[84].这或许可以为通过控制碳含量来调节材料的熔点提供一些思路.
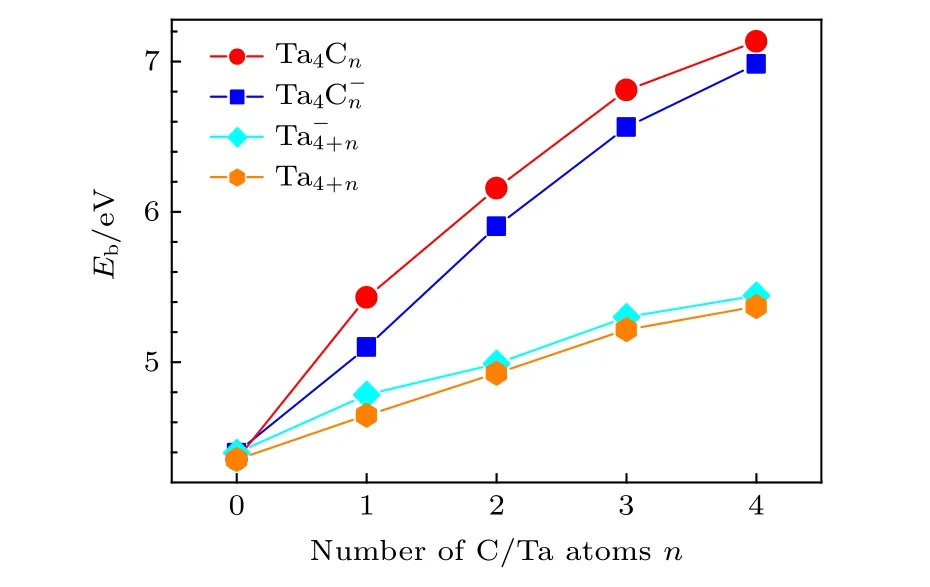
图8 (n = 0—4)团簇的单原子结合能(Eb)随碳/钽原子增加变化图Fig.8.Size-dependence of binding energies per-atom (Eb) of(n = 0–4) clusters.
表2 (n = 0—4)团簇的单原子结合能(Eb)Table 2.Binding energies per-atom (Eb) of and (n = 0–4) clusters.
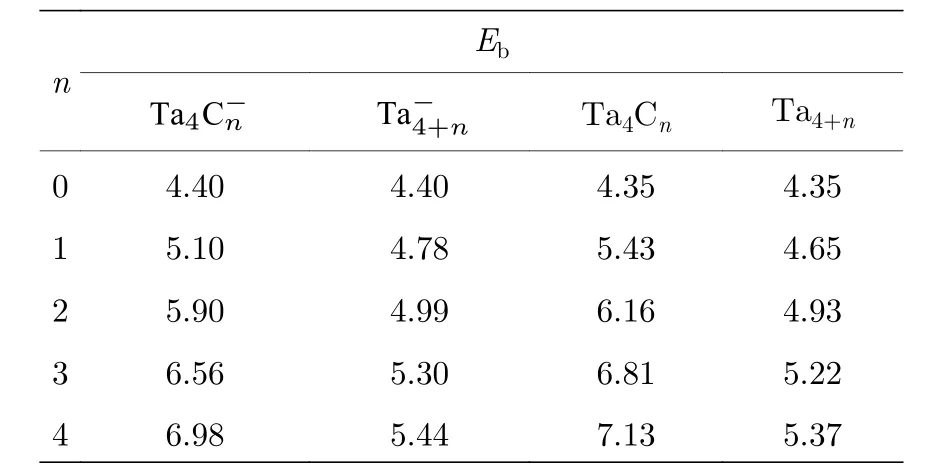
表2 (n = 0—4)团簇的单原子结合能(Eb)Table 2.Binding energies per-atom (Eb) of and (n = 0–4) clusters.
Eb n Ta4C-n Ta-4+n Ta4Cn Ta4+n 0 4.40 4.40 4.35 4.35 1 5.10 4.78 5.43 4.65 2 5.90 4.99 6.16 4.93 3 6.56 5.30 6.81 5.22 4 6.98 5.44 7.13 5.37
4 结 论
猜你喜欢
大学物理(2022年9期)2022-09-28
分析科学学报(2021年3期)2021-07-14
色谱(2021年6期)2021-05-06
物理通报(2020年7期)2020-07-01
科技资讯(2020年12期)2020-06-03
空天防御(2020年1期)2020-04-13
照明工程学报(2019年1期)2019-06-13
新课程研究(2016年1期)2016-12-01
原子与分子物理学报(2015年3期)2015-11-24
原子与分子物理学报(2015年3期)2015-11-24
