
Plackett-Burman联用Box-Behnken响应面法优化马来酸桂哌齐特脂质体的制备及表征*
2021-02-04 02:55张小雯孙敬蒙汪卓明宁劲涛王丹张炜煜
医药导报 2021年2期
张小雯,孙敬蒙,汪卓明,宁劲涛,王丹,张炜煜
(1.长春中医药大学药学院,长春 130117;2.吉林大学白求恩第一医院药学部,长春 130021)
近年来,心脑血管疾病的发病率逐年升高。心脑血管系统疾病是一种主要由血管壁病变,血液成分组成和血液动力学改变,以及不良生活习惯造成的一种常见的慢性疾病[1],多发生于中老年人。主要表现为高血压、心肌梗死、心绞痛与脑卒中、脑梗死等症状[2]。马来酸桂哌齐特(cinepazide maleate,CM)是新一代哌嗪类药物,在治疗心脑血管系统疾病方面获得国内外认可,目前上市品种仅有注射用粉针剂[3]。笔者在本研究以CM作为模型药物[4],由于CM遇光不稳定,在光照条件下含量急剧下降,半衰期为70 min。将CM制备成脂质体[5],增加药物稳定性,并进一步将脂质体混悬液固化干燥为粉末,改善其上述的缺点。笔者采用鱼骨图分析法[6],对影响CM制备因素进行剖析,结合Plackett-burman design(PBD)及Box-behnken design(BBD),优化马来酸桂哌齐特脂质体(cinepazide maleate liposomes,CM-Lip)的制备工艺,并进一步进行验证和表征,为新型药物的开发研究提供理论依据。
1 仪器与试药
1.1仪器设备 Agilent 1200高效液相色谱仪(美国安捷伦公司)、RE-52AA旋转蒸发器(上海亚荣生化仪器厂)、AR2140型电子天平(梅特勒-托利多仪器有限公司,感量:0.1 mg)、HH.S21-Hi6电热恒温水浴锅(北京市长源实验设备厂)、KQ2200型超声波清洗器(昆山市超声仪器有限公司)、透射电子显微镜(HITACHI公司)、PHS-3C型数字酸度计(仪电科学仪器)、SZ516 红外分析光谱仪(北京北分瑞利)、DSC3差示扫描量热仪(梅特勒-托利多仪器有限公司)、LS13320型激光粒度分析仪(江苏省金坛市荣华仪器制造有限公司)。
1.2材料与试剂 CM对照品(中国食品药品检定研究院,批号:100135-201105,含量:99.91%)、CM原料药(吉林省博大伟业制药有限公司,批号:171118,含量:98.97%)、胆固醇(北京鼎国昌盛生物科技有限责任公司,批号:DH061-1.1)、大豆磷脂(上海太伟药业股份有限公司,批号:201712061),甲醇、乙腈均为色谱级由Sigma公司提供,磷酸二氢钠(批号:20170617)、磷酸氢二钠(批号:20180215)、磷酸二氢钾(批号:20180103)、磷酸氢二钾(批号:20180402)均购自北京化工厂。
2 方法与结果
2.1含量测定方法
2.1.1色谱条件 采用Aglient-XDB C18色谱柱(4.6 mm×250 mm,5 μm);流动相:乙腈-0.05 mol·L-1磷酸氢二钠缓冲液(25:75);柱温:30 ℃;流速:1.0 mL·min-1;进样量:20 μL;检测波长:230 nm。
2.1.2溶液的制备
①对照品溶液的制备。取CM对照品2 mg,精密称定,置于10 mL量瓶中,加流动相溶解并定容,摇匀,滤过,即得。
②供试品溶液的制备。取CM-Lip 混悬液,置于250 mL锥形瓶中,加PBS(pH值=5.8)稀释至刻度,备用;取PBS(pH=7.4) 8 mL置透析袋中,将透析袋置于上述锥形瓶中,并放置(37±1℃)℃,75 r·min-1恒温振荡器中,振荡180 min后,从透析袋中取出样品5 mL,置于蒸发皿内放置水浴锅上蒸干,残渣加流动相溶解并定容至5 mL,摇匀,滤过,即得。
③阴性对照溶液的制备。取磷酸氢二钠0.709 8 g,加水溶解成500 mL,配成0.05 mol·L-1的磷酸氢二钠溶液,备用;取乙腈25 mL置于100 nL量瓶中,加上述备用液定容至刻度,摇匀,滤过,即得。
2.1.3专属性实验 精密量取对照品溶液,供试品溶液,阴性对照溶液20 μL,按照“2.1.1”项色谱条件进样测定。结果供试品溶液中CM成分得到较好分离,供试品与对照品在相同的保留时间出现色谱峰,而阴性色谱图中在对照品相同保留时间处没有色谱峰,证明阴性无干扰,专属性良好。见图1。
2.1.4标准曲线的制备 分别精密吸取CM对照品溶液0.02,0.10,0.20,0.40,0.50,1.00 mL,置2 mL量瓶中,加流动相溶解并稀释至刻度,摇匀,滤过,取续滤液,即得不同浓度对照品溶液。依次以“2.1.1”项色谱条件进样测定,记录色谱峰面积,以色谱峰面积(Y)为纵坐标,对照品浓度(X)为横坐标,得回归方程:Y=14.375X-62.819(R2=0.999 9),表明对照品浓度在4.24~169.6 μg·mL-1范围内线性关系良好。
2.1.5精密度实验 取同一供试品溶液,重复进样测定6次,记录峰面积,计算峰面积RSD值为0.47%(n=6);表明测定方法精密度良好。
2.1.6稳定性实验 取同一供试品溶液,在0,2,4,6,12,24 h进样测定,计算CM供试品溶液峰面积平均值为1014.78,RSD值为0.94%(n=6),表明CM供试品溶液在24 h内保持稳定。

A.阴性样品;B.对照品;C.供试品;1.马来酸桂哌齐特。
2.1.7准确度实验 按处方量120%、100%、80%,精密称取CM,并按处方比例加入其他辅料,制备3种不同浓度的CM溶液,平均回收率分别为98.71%,99.70%,98.96%,RSD值分别为0.70%,1.58%,0.35%(n=3)。结果表明,均符合《中华人民共和国药典》2015年版四部通则9101项下规定。
2.2工艺过程 取磷脂、胆固醇溶解于有机溶剂中形成油相,取CM溶解于磷酸盐缓冲液(PBS)中形成水相,将水相缓慢滴加到油相中,短时间间歇超声,至形成W/O型乳剂;减压回收溶剂,至胶态,加PBS溶液,水合,再减压回收溶剂,即得CM-Lip混悬液。
2.3包封率的测定 采用反透析法[7],取CM-Lip混悬液,置于250 mL锥形瓶中,加PBS(pH值5.8)稀释至刻度,备用;取PBS(pH值=7.4)8 mL溶液置透析袋中,将透析袋置于上述锥形瓶中,并放置(37±1)℃,75 r·min-1恒温振荡器中,振荡180 min,从透析袋中取出样品5 mL,蒸干,加流动相溶解,置于5 mL量瓶,稀释至刻度,经孔径0.45 μm微孔滤膜滤过,取续滤液,按“2.1.1”项下色谱条件测定,计算包封率(EE),EE(%)=(C总-C游离)/C总×100%。式中C总为药物总量,C游离为游离药物总量。
2.4PBD PBD是一种在影响因素较多时,可以用较少实验次数科学、高效的筛选出显著影响因素的两水平实验设计方法[8]。根据预实验结果综合分析,并运用鱼骨图(图2)分析可知,影响CM-Lip包封率的因素包括处方、方法、设备、环境四大项,共23种影响因素;通过预实验,确定有机溶剂用量(A)、有机溶剂(乙醚:乙醇)比例(B)、水合温度(C)、超声时间(D)、旋蒸温度(E)、磷脂与药物比(F)、水合时间(G)、油相(乙醚:乙醇)与水相比例(H)为主要影响因素。采用Minitab 17.0软件进行Plackeet- Burman设计,以主要影响因素为自变量,设定各因素两水平(-1、+1)的取值,以包封率为因变量,设计因素水平设计见表1。

图2 包封率的影响因素的鱼骨图
实验结果见表2,用Minitab 17.0软件进行数据分析处理,得到其回归模型为:EE=79.6-0.135A-2.92B+0.093C+0.067D-0.487E+1.127F+4.73G+4.77H;通过对实验数据进行多元回归分析,结果见表3,模型P值为 0.04,F值为7.54,差异有统计学意义(P<0.05);r=0.986 5,提示回归拟合程度较好。由图3a、3b可知,F、G及H差异有统计学意义(P<0.01),对CM-Lip包封率显著性排列为F>H>G;A、B、C、D、E对包封率无显著性影响(P>0.05)。在水合过程中,水合温度的变化能够显著影响CM-Lip的包封率,若水合温度过高,可能导致CM-Lip的稳定性有所下降,出现CM渗漏,CM-Lip包封率下降的现象。加入一定比例的油相(乙醚:乙醇)与水相,但当其比例较大时,超声过程中会出现分层现象,会使CM-Lip的包封率下降,应使油相(乙醚:乙醇)与水相比恰当,进一步提高CM-Lip的包封率。当磷脂与药物比高于最佳比例时,存在包封率下降的现象,当磷脂与药物比过低时又使得空白脂质体大量存在。
2.5响应面设计实验 响应面法是指通过一定数量实验次数对各个实验因素及其因素之间的相互作用进行分析,最终得到直观三维曲面图用以进行评价的优化方法[9-10]。在PBD实验的基础上,结合鱼骨图分析,最终选择磷脂与药物比例、水合时间、油相(乙醚:乙醇)与水相比例作为Box-Behnken响应面实验的3个主要因素,每个因素分为3个水平,因素水平设计表见表4,根据Design-Expert.V8.0.6.1 软件设计表进行实验。以包封率为响应值,采用Box-Behnken响应面实验法对3个因素进行优化,实验结果见表5。

表1 Plackeet-Burman设计因素水平

表2 Plackeet-Burman实验结果

表3 Plackeet-Burman实验设计的显著性结果
应用Design-Expert.V8.0.6.1对表5进行分析,结果见表6,以包封率对各因素自变量进行模型拟合,通过拟合得到的二次回归方程为Y=75.51+9.93×A+9.50×B-0.65×C-3.85×A×B-7.98×A×C-1.35B×C-13.15×A2-13.75×B2-0.16×C2(R2=0.950 6,失拟度检验F值为5.63,P=0.064 2>0.05),由拟合方程结果可知拟合模型F值为14.98(P<0.05),说明拟合模型拟合度高,失拟项F值较小为5.63,其P值为0.064 2>0.05,表明其失拟项不显著性,表明该拟合模型拟合结果良好,可用于实验结果分析,信噪比值为14.274,相对较高,证明该模型可用于实验结果分析;二次回归拟合方程的决定系数R2=0.950 6>0.95,证明实测值与预测值之间的相关性较高,能够高度准确的预测实际情况。
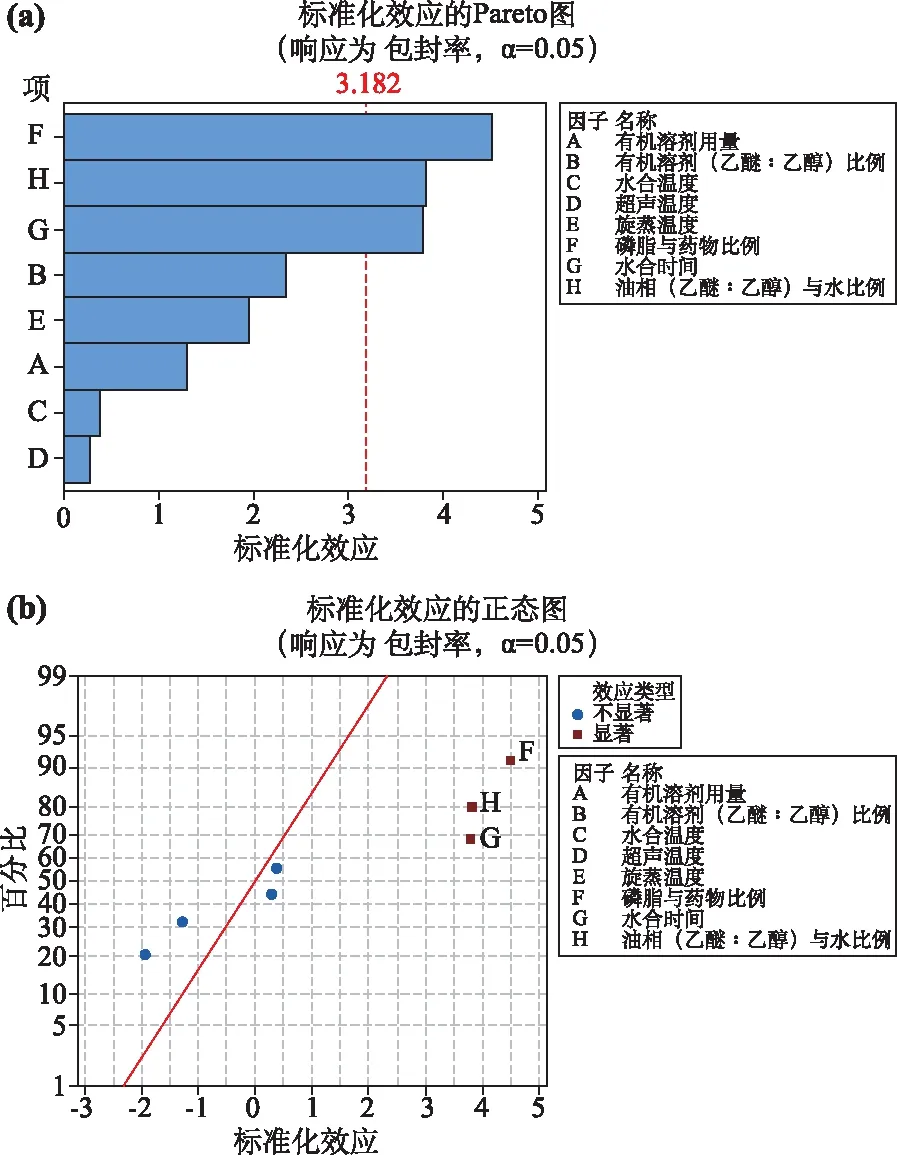
图3 响应值(Y)的Pareto图(a)及正态图(b)

表4 Box-Behnken设计因素水平设计

表5 Box-Behnken实验设计与结果

表6 Box-Behnken设计方差分析结果
根据二次回归方程拟合结果,将其中一个因素水平固定,进行EE对其他两个因素的拟合,并得到实验结果的等高线与三维效应曲面图对CM-Lip的制备工艺参数进行优选,图4~6中包封率随着各变量的增加而增加,到达最大值后逐渐下降。软件的处方优化的结果为:药物与磷脂比例为1:10、油相(乙醚:乙醇)与水相比例为3:1,水合时间为1.5 h。

图4 因素A及因素B对综合评分EE的等高线图和三维响应面图

图5 因素A及因素C对综合评分EE的等高线图和三维响应面图
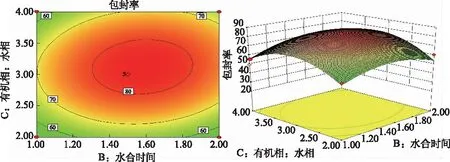
图6 因素B及因素C对综合评分EE的等高线图和三维响应面图
2.6验证实验 根据响应面实验结果的最优工艺,即药物与磷脂比例为1:10、油相(乙醚:乙醇)与水相比例为3:1,水合时间为1.5 h,预测包封率为84.32%,进行验证,结果,3批CM-Lip包封率分别为84.41%,79.16%,85.79%,平均包封率为83.12%,RSD值为0.22%,偏差较小,预测性良好,证明脂质体最佳制备工艺的稳定可行性。
2.7CM-Lip粉末的表征验证
2.7.1脂质体的形态 在最优工艺处方条件下,取“2.2”项下制备的CM-Lip混悬液,以纯化水适当稀释后用2%磷钨酸负染,滴至专用铜网上,自然挥干,静置使粒子在铜网上沉积,放大倍率200 000倍,加速电压100 kV,观察并摄像。图7结果显示,CM-Lip的外观呈不规则球形或椭球形。

图7 马来酸桂哌齐特脂质体的透射电镜图(×200 000)
2.7.2粒径分布与Zeta电位 在最优工艺处方条件下,取“2.2”项下制备的CM-Lip混悬液,采用激光粒度分析仪测定粒径及分布,结果见图8。CM-Lip混悬液粒径为(114.5±10.3) nm,分散系数(PDI)为0.208,说明CM-Lip混悬液粒径分布较均匀;Zeta电位为-17.85 mV,绝对值>15 mV,表明CM-Lip混悬液稳定,不易发生絮凝。

图8 马来酸桂哌齐特脂质体粒径分布
2.7.3CM-Lip载药量的测定 载药量是CM-Lip中所含药物量的百分率[11]。有机试剂破坏法是比较常见并且容易的测定载药量的方法。取“2.2”项下制备的CM-Lip混悬液,加甘露醇搅拌使溶解,冷冻干燥得CM-Lip粉末。精密称取CM-Lip粉末0.60 g,用PBS(pH值7.4)充分溶解,置于10 mL量瓶中并稀释至刻度,摇匀,分别精密量取摇匀后溶液1 mL,置于不同离心管中,向离心管中分别加入甲醇、异丙醇、无水乙醇及其不同比例的混合溶剂6 mL,超声、离心破坏,精密吸取离心后上清液1 mL,蒸干,残渣加流动相溶解并定容至2 mL,摇匀,经孔径0.45 μm微孔滤膜过滤,取续滤液,按“2.1.1”项下色谱条件测定,计算载药量。载药量(%)=脂质体中所含药量/脂质体总量×100%。3次测定结果分别为30.1,30.5,29.9 mg·g-1,平均载药量为30.17 mg·g-1,RSD=1.01%。
2.7.4差示扫描量热(differential scanning calorimetry,DSC)分析 取CM、胆固醇、甘露醇、物理混合物与“2.2”项下制备的CM-Lip混悬液,加甘露醇搅拌使溶解,冷冻干燥得CM-Lip粉末,进行DSC分析。工作条件:以空铝坩为空白参考池,另一铝坩为样品池,在样品池中放入样品约5 mg,置于机器中进行图谱扫描;N2作为吹扫气,扫描速度50 ℃·min-1,扫描范围30~300 ℃。图9结果表明,CM在177.94 ℃时出现一个熔点峰,甘露醇在167.04 ℃出现一个熔点峰,胆固醇在153.65 ℃时出现一个熔点峰,但是当药物与两者物理混合得到物理混合物a后,测定时却只出现了两个峰,说明是药物峰与甘露醇峰发生了重合,因此制备不含甘露醇的物理混合物,得到了物理混合物b;CM-Lip的DSC出现新的熔点峰,且峰值发生变化,说明形成新物质,不是简单地物料叠加,证明生成了CM-Lip。

a.马来酸桂哌齐特;b.甘露醇;c.胆固醇;d.马来酸桂哌齐特脂质体;e.物理混合物a;f.物理混合物b。
图9 DSC脂质体总图
a.cinepazide maleate;b.mannitol;c.cholesterol;d.cinepazide maleate liposomes;e.physical mixture a;f.physical mixture b.
Fig.9 General map of DSC liposomes
2.7.5红外光谱扫描 分别取CM、胆固醇、物理混合物与“2.2”项下制备的CM-Lip混悬液,加甘露醇搅拌使溶解,冷冻干燥得CM-Lip粉末。加入适量溴化钾固体,充分研细,装片,压片,在400~4000 cm-1范围内进行红外分光扫描,比较各谱图曲线,见图10。图10A显示,1644 cm-1处吸收峰为胆固醇的C=C伸缩振动峰,3 404 cm-1处的吸收峰为胆固醇的-OH伸缩振动峰,2 931cm-1为-CH3的伸缩振动峰,1354~1460 cm-1为-CH3的不对称伸缩振动。图10C显示,3 450 cm-1处吸收峰

图10 胆固醇(A)、物理混合(B)、马来酸桂哌齐特(C)、马来酸桂哌齐特脂质体(D)的红外光谱图
为CM的-OH伸缩振动峰,2 590 cm-1处吸收峰为哌嗪环叔胺盐的N-H伸缩振动峰,1244和1121 cm-1处吸收峰分别为C-O-C的不对称伸缩振动和对称伸缩振动,1633,1589和1499 cm-1处吸收峰为苯环的骨架伸缩振动,2976和2875 cm-1分别为CH3的-CH-不对称伸缩振动和对称伸缩振动。图10 B显示具有各个简单物质胆固醇和CM的色谱峰。由图10 C和图10D显示,当CM被制备成脂质体以后,-CH3的不对称伸缩振动、哌嗪环叔胺盐的N-H伸缩振动和C-O-C不对称伸缩振动几乎没有变化,说明CM进入脂质体囊泡中。此外,CM在3450 cm-1处-OH伸缩振动峰吸收峰消失,在3400 cm-1处形成宽峰,进一步说明CM进入脂质体囊泡中。
2.8释放度实验 采用体外透析法测定CM-Lip的体外释放过程,分别取CM原料药混悬液和与CM-Lip混悬液,分别置于250 mL量瓶中,并加PBS(pH值=5.8)稀释定容至刻度,转移至250 mL锥形瓶中;分别放入5个含有PBS(pH值=7.4)溶液8 mL的透析袋,置于(37±1)℃、75 r·min-1的恒温振荡器中振荡(仪器为不透光装置),分别于0.5,2,4,6,12 h将取一个透析袋,精密吸取溶液5 mL,取样时以暗箱为转移工具,使样品溶液保持避光条件,并补充相同温度PBS(pH值=7.4)的释放介质8 mL,蒸干,残渣加乙腈-0.05 mol·L-1磷酸二氢钠缓冲液溶解并稀释定容至5 mL,摇匀,经孔径0.45 μm微孔滤膜滤过,取续滤液,按“2.1.1”项下色谱条件测定,计算释放度,在相同条件下做平行实验3次,药物累计释放度计算绘制药物累计释放曲线,并进行曲线拟合,结果见图11。结果表明,在同一释放介质中,CM原料药0.5 h达到80%,而CM-Lip释放度为30%。CM-Lip始终保持缓慢释放,12 h累积释放度为76%,证明CM-Lip具有明显的缓释作用,解决CM半衰期短的缺点。将CM-Lip释放曲线分别用零级、一级和Higuchi方程进行拟合,结果显示,拟合方程及相关系数(r)分别为Q=0.062 7t+0.315 7(r=0.974)、Q=0.701 6(1-e-0.752 4t)(r=0.967)、Q=0.254 1t1/2+0.213 2(r=0.976),Q为累积释放度,t为时间。根据相关系数判断,结果表明其释药行为符合Higuchi方程。
3 讨论
脂质体的主要组成材料为磷脂和胆固醇等,在水中能自发形成具有双分子层结构的封闭囊泡(vesicles)[12]。实验过程中发现,仅采用逆向蒸发法制备的CM-Lip包封率较低,主要原因是该方法水合时间较长,易出现水合不完全。此外,逆向蒸发法不容易控制脂质体的粒径,因此通过逆向蒸发法制备CM-Lip混悬液后,再通过高压均质机,可使脂质体粒径减小且均匀,而且通过高压均质机的挤压作用后进一步提高了其包封率,包封率最高为84.9%。
包封率常用来做评价脂质体制备工艺优劣的生物重要指标。常见测定包封率的方法有透析法、超速离心法、凝胶过滤法等。因凝胶过滤法过程复杂消耗时间以及成本较高,但是采用超速离心法离心转速比较高,在离心的过程中造成脂质体中药物渗漏,不能确保脂质体中游离药物的分离是否完全,从而导致脂质体的包封率较低,结果不准确;故本实验考察透析法(正透析与反透析)与超速离心法分法测定CM-Lip的包封率,预实验结果表明,正透析实验结果中游离药物一直难以达到平衡,反透析实验结果中药物平衡的稳定性良好,在透析30~100 min,CM含量在一直逐步增加,150~200 min药物游离量趋于平衡。故最终选择反透析法测定CM-Lip包封率。乳剂的形成是逆向蒸发法制备脂质体的关键因素之一,前期预实验表明有机相溶剂为乙醚与乙醇,且比例为3:1时,脂质体包封率最大;当乙醚与乙醇的比例>3:1时,油相(乙醚:乙醇)与水相会分层,不会形成乳剂;当乙醚与乙醇的比例<3:1时,油相(乙醚:乙醇)与水相不分层,但包封率较低,因此最终选择有机相溶剂乙醚与乙醇比值为3:1。

图11 马来酸桂哌齐特原料药混悬液和马来酸桂哌齐特脂质体的体外释放曲线
本研究采用鱼骨分析法,对影响CM-Lip制备因素进行剖析,结合PBD及BBD,优化CM-Lip的制备工艺。传统的正交实验设计利用线性数学模型,分析出多个因素水平的最佳组合;但正交设计只能分析离散型数据,具有预测性不佳的缺点。PBD结合BBD,采用线性模型,能求得回归方程,通过合理预测得出最佳工艺条件。
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13
中国美容医学(2022年3期)2022-04-27
昆明医科大学学报(2022年3期)2022-04-19
四川大学学报(自然科学版)(2022年1期)2022-02-10
健康之家(2021年19期)2021-05-23
中国药学药品知识仓库(2021年18期)2021-02-28
信息技术时代·上旬刊(2019年4期)2019-09-10
健康大视野(2018年10期)2018-10-29
科技风(2018年10期)2018-05-14
分析化学(2015年8期)2015-08-13
