
茶树DNA去甲基化酶基因的全基因组鉴定及表达分析
2021-02-06 06:35陈瑶张伟富任恒泽熊飞张豪杰姚利娜刘莹王璐王新超杨亚军郝心愿
茶叶科学 2021年1期
陈瑶,张伟富,任恒泽,熊飞,张豪杰,姚利娜,刘莹,王璐,王新超,杨亚军,郝心愿
茶树DNA去甲基化酶基因的全基因组鉴定及表达分析
陈瑶,张伟富,任恒泽,熊飞,张豪杰,姚利娜,刘莹,王璐,王新超,杨亚军*,郝心愿*
中国农业科学院茶叶研究所/国家茶树改良中心/农业农村部茶树生物学与资源利用重点实验室,浙江 杭州 310008
DNA去甲基化酶(dMTase)是主动去甲基化过程中具有糖基化酶和脱嘌呤裂合酶活性的双功能酶。基于茶树基因组数据,本研究利用生物信息学方法对茶树DNA去甲基化酶()编码基因进行了全面鉴定和序列分析。全基因组鉴定结果表明,舒茶早基因组中包含4个,系统进化分析将分为和两个分支,基因结构分析显示不同成员之间的基因序列长度和内含子数量存在差异。不同蛋白的理化性质相似,均包含ENDO3c和RRM_DME典型的保守结构域,且都定位在细胞核中。家族基因启动子区域包含大量光信号响应、植物激素响应、胁迫响应和生长发育等相关顺式作用元件。表达分析结果显示,e基因在不同组织器官中均有表达,有一定的组织特异性;在炭疽菌()、拟盘多毛孢菌()和茶尺蠖()等生物胁迫及干旱等非生物胁迫下,的表达均被显著诱导;冬季休眠过程中在不同品种成熟叶和越冬芽中的表达模式存在显著差异;在花芽发育及种子萌发的不同阶段的表达存在显著上调过程。CsdMTases介导的主动去甲基化过程在调控茶树胁迫应答和生长发育中可能发挥了重要的作用,为深入揭示茶树表观调控机制提供理论依据。
茶树;DNA去甲基化酶;逆境胁迫响应;生长发育;表达分析
DNA甲基化是表观遗传调控的重要机制之一,在调控基因表达、基因印记、转座子沉默以及维持基因组稳定性中发挥重要作用。在植物中,DNA甲基化主要通过DNA甲基转移酶将S-腺苷甲硫氨酸(-adenosyl methionine,SAM)的甲基基团转移到胞嘧啶的第5位碳原子上,形成5-甲基胞嘧啶(5-methylcytosine,5-meC)[1]。依据甲基化的过程,可以大致分为2种类型:(1)从头合成甲基化,即两条链均未被甲基化的DNA被甲基化;(2)维持甲基化,以甲基化的DNA单链为模板,通过DNA复制对新生链进行甲基化修饰。依据甲基化的碱基位点,可以将其分为3种类型:CG、CHG、CHH(H指A、C或T)。在植物中,CG位点的甲基化由甲基转移酶(Methyltransferase 1,MET1)维持[1];对称的CHG的甲基化主要由染色质甲基化酶(Chromomethylase 3,CMT3)与组蛋白抑制标记结合对未甲基化的CHG进行修饰[1];而非对称的CHH的甲基化的建立主要通过RNA介导的从头合成途径(RNA-directed DNA methylation,RdDM)完成[2]。典型的RdDM包含3个主要的步骤:RNA聚合酶Ⅳ的转录,从头合成的DNA甲基化和异染色质的形成[3]。植物中3种类型的甲基化均会引起植物整体DNA甲基化水平的升高。基因组中DNA甲基化的水平不仅取决于DNA甲基化的建立与维持,也取决于DNA去甲基化的发生。DNA去甲基化是DNA甲基化的相反过程,即5-甲基胞嘧啶被未修饰的胞嘧啶替代的过程。在植物中,DNA去甲基化可以分为2类,一类是被动的去甲基化,是由于DNA复制过程中甲基转移酶活性的丧失或甲基供体的缺乏导致甲基化无法维持,从而使整个基因组DNA甲基化水平降低[4];另一类是主动的去甲基化,即不依赖DNA复制,由一系列酶催化的酶促反应过程。与DNA甲基化不同的是,去甲基化的完成需要包含DNA糖基化酶/裂合酶(DNA glycosylase/lysase)在内的一系列酶的催化,而不是单一的DNA甲基转移酶。在植物DNA主动去甲基化过程中,DNA糖基化酶/裂合酶可以识别并直接去除5-meC。DNA糖基化酶通过特异性裂解N-糖基键切除5-meC[5],从而启动碱基切除修复机制。随后,裂合酶对DNA进行切割,产生缺乏核苷酸的位点,并在切口处产生3′-羟基,DNA聚合酶向其添加未甲基化的胞嘧啶,DNA连接酶通过密封切口来完成修复过程。因此,基因组DNA甲基化的总体水平受DNA甲基转移酶和DNA去甲基化酶(DNA demethylase,dMTase)的共同作用。
dMTase是主动去甲基化过程中具有糖基化酶和脱嘌呤裂合酶活性的双功能酶,含有1个具有DNA结合活性的典型的双螺旋发卡结构,主要负责识别并移除5-mC碱基、裂解DNA骨架,以便未甲基化胞嘧啶的添加。植物5-meC DNA糖基化酶归属DEMETER-LIKE(DML)家族,是分子量最大、功能最丰富的DNA糖基化酶。在拟南芥中已鉴定的去甲基化酶有ROS1(Repressor of silence 1),DME(Transcriptional activator demeter),DML2(Demeter-like protein 2)和DML3(Demeter-like protein 3),4个dMTase均归属DNA糖基化酶家族,除包含1个典型的由HhH-GPD(Helix–hairpin–helix and a Gly/Pro rich loop)组成的具有催化活性的糖基化酶结构域外,还在氮端含有1个短的富含亮氨酸的结构域以及碳端含有1个对催化活性必不可少的大结构域[6]。研究表明,DME可以激活被甲基化沉默的印迹基因的母体表达,对拟南芥胚乳建立基因组印迹至关重要[7]。ROS1可以通过去甲基化目标DNA启动子来抑制同源依赖的转录基因沉默,是释放高甲基化转基因的转录沉默所必需的糖基化酶[8]。DML2和DML3可以通过去除基因5′和3′甲基化,从而保护内源基因免受潜在的甲基化危害[5]。在其他植物中,已鉴定番茄()中含有基因3个[9],水稻()中6个[10],杨树()中5个[11]。尽管在上述植物中基因已得到广泛分析和鉴定,但是的保守性及多态性显示不同植物、不同物种中的数量和功能都存在很大差异,而相对于模式草本植物而言,对多年生木本植物中家族基因的研究还有待深入。
茶树是多年生木本经济作物,分布地域广,生长周期中会遇到低温、干旱、病虫害等生物和非生物逆境。茶树在冷驯化阶段,其基因组伴随着DNA甲基化水平的变化,表明DNA甲基化参与茶树的冷驯化过程,与茶树的抗寒性息息相关[12]。DNA甲基化酶和基因在干旱胁迫过程中相对表达量呈现显著变化[13]。当前有关茶树DNA去甲基化酶()的研究还不够深入,在茶树的生长发育以及生物胁迫调控中的作用尚不清楚。因此,本研究以公布的小叶种茶树舒茶早基因组为参考,对茶树家族基因进行了全面的鉴定。结合表达分析,以期揭示在茶树生长发育(开花、种子萌发、芽休眠形成及解除)和逆境胁迫,如炭疽菌()、拟盘多毛孢菌()、茶尺蠖()和干旱中的作用。
1 材料与方法
1.1 试验材料及处理
以种植于中国农业科学院茶叶研究所试验茶园的20年生茶树舒茶早、龙井43和碧云为试验材料,分别采取茶树的幼嫩叶片和顶芽用于的鉴定。
以种植于中国农业科学院茶叶研究所试验茶园的20年生茶树早生品种龙井43,中生品种碧云,晚生品种政和大白为试验材料,从2018年9月下旬茶树进入休眠期开始至翌年休眠解除止,依据芽的发育和天气情况于茶树休眠形成阶段、深休眠阶段和萌发阶段采摘剪口下第一腋芽和成熟叶,用于茶树芽和叶休眠形成及解除阶段的表达分析。
以龙井43为试验材料,采取自然生长条件下茶树的根、茎、叶、顶芽、腋芽、花蕾和盛花用于组织特异性表达分析;参照熊飞等[14]所述的生物胁迫取样方法,以未接种茶树为对照,给茶树分别接种炭疽菌、拟盘多毛孢菌、茶尺蠖处理,于12 h和24 h采取接种后的叶片用于茶树生物胁迫下的表达分析;于2019年11月上旬采取茶树不同阶段的花用于花芽发育阶段基因表达分析,同时采回种子用于茶树种子萌发过程的表达分析,处理方法为将种子均分为两份,一份作为萌发前基因表达量测定,另一份扒掉种皮后埋于珍珠岩中萌发,待胚根长至1~2 cm时拔出洗净用于萌发后基因表达量测定。干旱胁迫取样以盆栽龙井43为材料,人工气候室正常培养(25℃,12 h光照/12 h黑暗,湿度65%),对照组正常浇水,处理组不浇水,处理18 d;每隔3 d取样1次,每次采取每盆的第2~3片叶子,用于基因表达分析。每次采样完毕对照组浇水。以上取样及处理均设3个生物学重复,取样后立即放入液氮,置于–80℃冰箱中保存备用。
1.2 家族基因鉴定
从拟南芥信息资源数据库(https://www.arabidopsis)下载拟南芥中已知的dMTase基因序列,并利用这些序列从茶树参考基因组[15-17]中通过Blast同源比对得到茶树舒茶早中的基因。为了进一步验证这些序列在茶树中的准确性,利用Pfam数据库31.0中的隐式马尔可夫模型(HMM)搜索,进一步验证了dMTase中两个特征结构域的存在。然后,检索已鉴定成员的基因组,转录本,编码序列和蛋白序列,利用ProtParam分析程序预测蛋白分子量、等电点、氨基酸组分等。之后,设计荧光定量引物用于基因短片段的克隆验证,PCR产物送至杭州有康生物科技有限公司进行测序,测序结果与原序列进行比对。
1.3 基因序列、结构比较分析
为了进一步了解茶树基因结构特征,从Phytozome(www.phytozome.net),NCBI(www.ncbi.nlm.nih.gov)和JGI(www.jgi.doe.gov)网站下载5种植物(水稻、玉米、葡萄、杨树、拟南芥)的dMTase蛋白序列。利用clustalw进行多序列比对,采用邻接法利用MEGA 7.0进行系统进化树构建。利用SMART对CsdMTase进行保守结构域分析。使用Tbtools软件对基因的进化树和保守结构域进行分析。利用Gene Structure Display Server 2.0(GSDS)进行基因内含子-外显子结构分析。为了分析启动子的顺式作用元件,从茶树基因组数据集中检索起始密码子的上游序列(2 000 bp),利用Plant CARE(http://bioinformatics.psb.ugent.be/webtools/plantcare/html)工具分析启动子顺式作用元件。分析结果使用Tbtools软件进行可视化。
1.4 核酸提取及表达分析
1.4.1 总RNA的提取及cDNA的合成
以龙井43、碧云、政和大白不同休眠阶段的越冬芽、成熟叶,龙井43、碧云和舒茶早幼嫩叶片,龙井43不同开花阶段的花、不同萌发状态的种子、干旱胁迫处理的叶片、生物胁迫处理的叶片为材料,按照CTAB法提取不同处理样品的总RNA,用NanoDrop ND2000 微量核酸蛋白测定仪测定RNA浓度,1%变性琼脂糖凝胶电泳检测总RNA质量,参照PrimeScript®RT reagent Kit反转录试剂盒[Takara,宝生物工程(大连)有限公司]说明书反转录合成cDNA用于基因表达分析。
1.4.2 基因表达分析
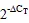
2 结果与分析
2.1 CsdMTase家族基因鉴定及理化分析
基于同源分析,从舒茶早基因组中鉴定出4个基因(CSS0047502.1、TEA031023.1、TEA022110.1、TEA033283.1),经过NCBI-blast与其他物种dMTase蛋白序列比对,将4个基因分别命名为和基因(表2)。对鉴定出的基因进行了基本特征分析发现,基因编码的氨基酸数目从1 800至1 943个不等,平均长度约为1 865个氨基酸;预测分子量为200.66~216.76 kDa,平均分子量约为208.49 kDa;理论pI为6.37至8.03,除CsDMEa外,其他蛋白的pI均小于7.0,为酸性蛋白;蛋白不稳定系数均大于40,表明茶树中CsdMTase蛋白不稳定。亲水性(GRAVY)分析显示茶树中所有的CsdMTase蛋白都是亲水性蛋白。亚细胞定位结果显示,CsdMTase蛋白均定位于细胞核中(表2)。
以茶树舒茶早、龙井43和碧云的cDNA为模板,用表1列出的荧光定量引物为模板,对茶树基因片段进行克隆,结果如图1所示,在3个茶树品种中均有条带,测序结果显示克隆得到基因片段与舒茶早基因序列一致,表明文中鉴定到的4个基因确实存在于茶树中。
2.2 CsdMTase家族基因的遗传发育树及保守结构域
为探究茶树基因家族的进化关系,本研究利用拟南芥、葡萄、杨树、水稻和茶树的CsdMTase蛋白序列构建系统进化树。结果表明,植物中的基因可以分为和两个分支。类基因存在于所有双子叶植物中,但在单子叶植物中不存在(图2)。葡萄和拟南芥都只有1个基因,而茶树和杨树中基因却有2个。虽然基因存在于所有研究的植物中,但基因的个数在各个植物中不尽相同(1~6个),其中水稻中最多,为6个。
利用SMART对dMTase进行保守结构域分析(图2),结果表明,大多数dMTase基本包含4个共同保守结构域:ENCO3c结构域(ENCO3C domain,包含HhH-GPD domain),Perm-CXXC结构域(Permuted single zf-CXXC unit,Perm-CXXC domain),RRM_DME结构域(RR DME domain)以及FES结构域(FES domain)。前人对拟南芥的研究表明ENCO3c结构域行使糖苷酶功能,Perm-CXXC结构域介导DNA 结合活性[18]。Perm-CXXC结构域和RRM_DME结构域是dMTase特异性结构域,这两者与ENCO3c结构域共同参与dMTase活性。因此,在我们的分析中,编码由ENCO3c结构域和DME结构域组成的蛋白质的茶树基因被鉴定为。在茶树中,所有的均含有ENCO3c、RRM_DME和Perm-CXXC等3个结构域。与ROS分支相比,DME分支的序列保守性更强(图2)。

表1 茶树CsdMTases基因家族实时荧光定量引物
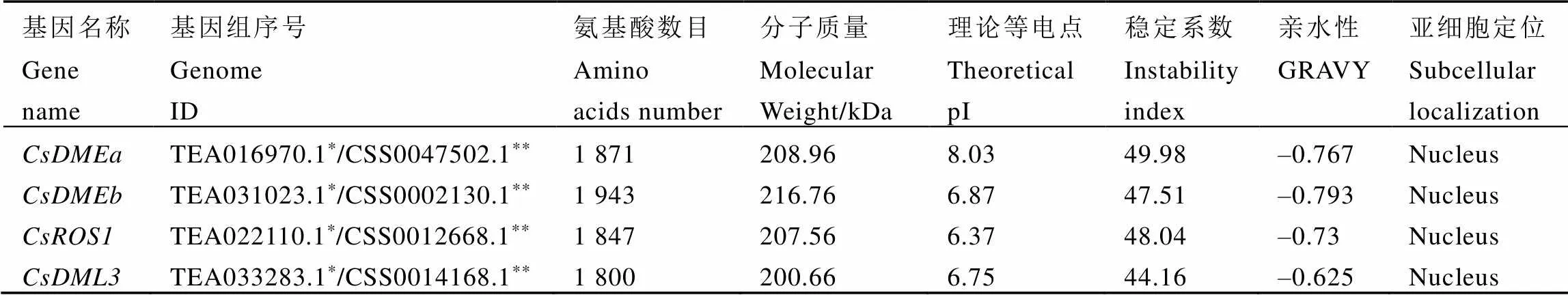
表2 茶树CsdMTases基因的基本特征
注:*是参考Wei等人[16]基因组;**是参考Xia等人[15]基因组
Note: * refers to the genome of Wei et al[16]. ** refers to the genome of Xia et al[15]
2.3 CsdMTase家族基因的结构及启动子分析
为进一步阐明茶树基因的结构特征,利用GSDS 2.0研究基因的外显子-内含子结构,分析显示基因包含15~19个外显子(图3-A)。其中,基因外显子数量最多,包含19个外显子,而和中都只含有15个外显子。的基因长度和的相似,都含有17个外显子。
利用Plant CARE数据库对基因上游2 000 bp顺势作用调控元件分析显示,基因上游含有大量顺式作用元件,大致可以分为4类:光响应、植物激素响应、胁迫响应以及植物生长发育相关的作用元件。其中,激素反应元件ABRE参与脱落酸反应,CGTCA-motif和TGACG-motif涉及茉莉酸甲酯响应,GARE-motif和P-box参与赤霉素响应,TCA-element参与水杨酸适应以及AuxRR-core和TGA-box参与生长素响应。启动子还具有多种应激反应调控元件,如参与低温应激的LTR,参与干旱诱导的MBS,涉及防御和应激反应的TC-rich repeats以及响应厌氧诱导的ARE调控元件。另外,还确定了一些组织特异性的顺式调控元件,例如参与种子特异性调控的RY-element、胚乳特异性表达所需的GCN4_motif以及与分生组织特异性激活有关的CAT-box。除此之外,还发现中含有参与玉米蛋白代谢调控的O2-site和与MYBHv1结合位点有关的CCAAT-box调控元件等。
总而言之,茶树中最常见的顺式调节元件主要与生理过程有关,例如光信号、胁迫应答和激素响应。茶树中多种顺式作用原件的存在表明基因可以参与茶树的生长发育以及胁迫应答。
2.4 CsdMTase的组织特异性分析
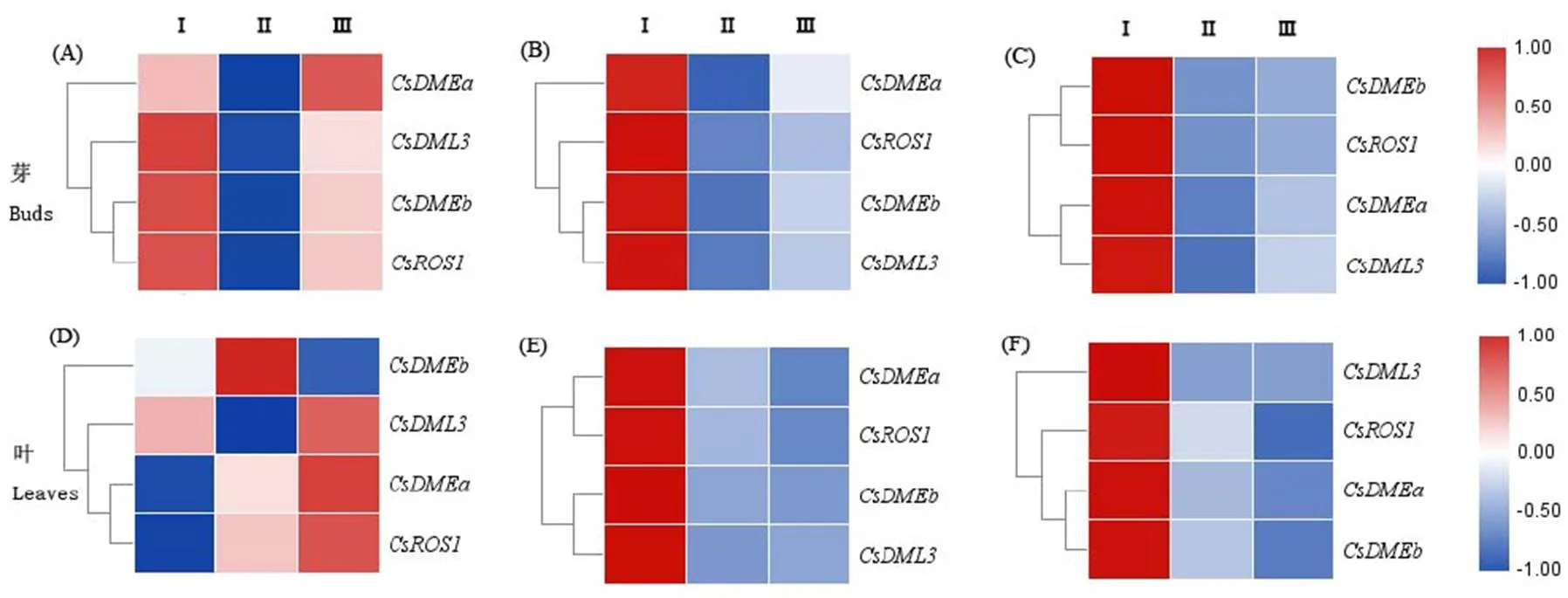
利用实时荧光定量PCR技术检测了在茶树龙井43的组织特异性表达。结果显示(图4),在花蕾、腋芽、茎中表达量较高,根中表达较低;在腋芽和根中表达差异显著;在不同组织的表达模式也不同,在花蕾中表达量最高,在腋芽和茎中的表达量较高,盛花、根和叶中的表达量较低。在花蕾和腋芽中的表达量较高,在盛花中基本不表达,而在其他组织均只有微弱的表达。
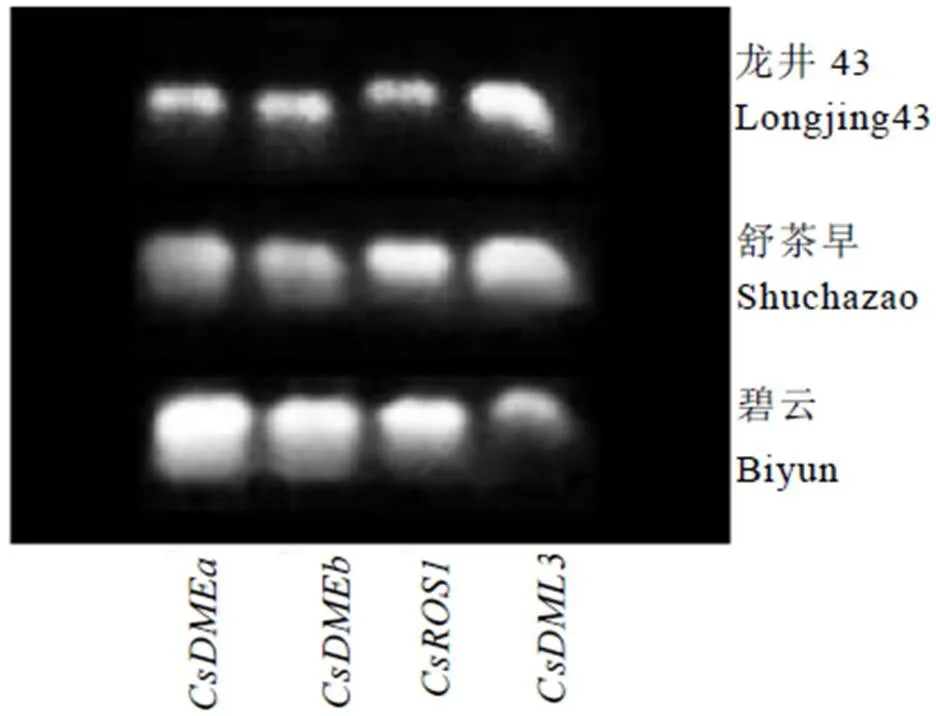
图1 茶树CsdMTases基因的鉴定

图2 CsdMTase进化树和保守结构域分析
2.5 CsdMTase在生物和非生物胁迫下的表达分析
为探究茶树在响应生物胁迫和非生物胁迫下的潜在作用,用实时荧光定量PCR技术分析了其在炭疽菌、拟盘多毛孢菌和茶尺蠖接种处理以及干旱、休眠诱导条件下的表达。茶树在接种炭疽菌处理12 h后,基因的所有成员表达量都达到最大值,并且与接种前相比均存在显著性差异,随后表达下调,至24 h时,、和的表达量仍较未处理前的高(图5),而和未处理前趋于一致(图5)。茶树在接种茶尺蠖处理时,除是在接种24 h才有显著性升高外,基因的表达模式也跟接种炭疽菌的一致。当给茶树接种拟盘多毛孢菌时,基因的表达模式与前两者有所差别。在接种拟盘多毛孢菌后,除外,基因的表达量持续上升,并且在24 h时达到最大。表明茶树受到生物胁迫时短期内能够诱导基因表达,该结果也和基因启动子包含与胁迫应激响应的顺式作用原件一致。

注:(A)茶树CsdMTase基因的内含子-外显子结构;(B)茶树CsdMTase启动子顺式调控元件
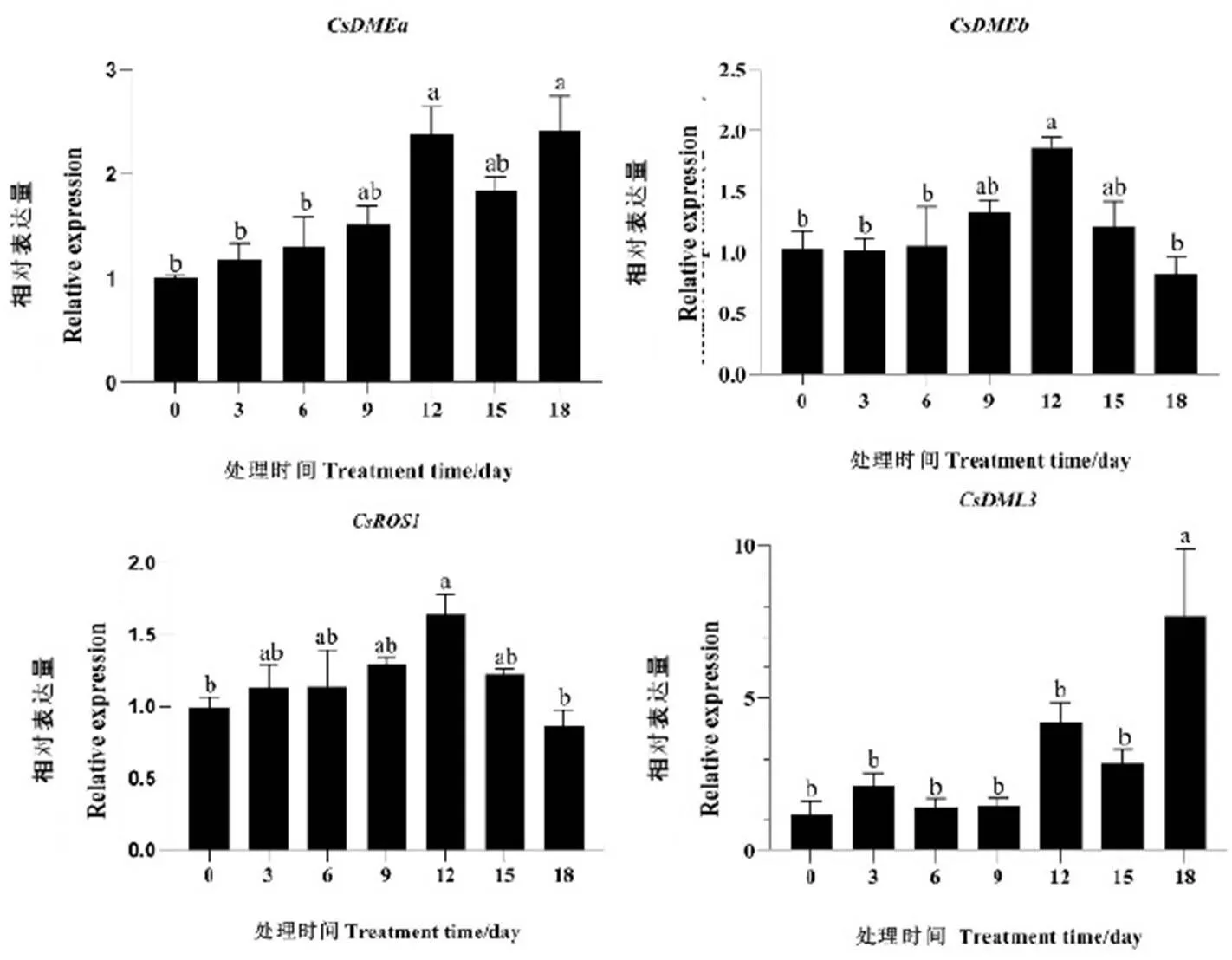
在干旱胁迫下,基因的所有成员都显示出转录表达量的显著变化(图6)。随着干旱处理时间的延长,基因的相对表达量也相应增加,除了外,基因的其余成员均在干旱处理12 d时相对表达量达到最大,并且与干旱处理0 d时有显著性差异。在干旱处理3 d时相对表达量有所增加,随着干旱处理时间的延长,基因的相对表达量下调,在12 d时又有所增加,并且在18 d时达到最大,表明基因可能在干旱胁迫过程中起到调控作用(图6)。
2.6 CsdMTase在生长发育过程中的表达分析
为探究在茶树生长发育中的可能调控作用,利用qRT-PCR技术分析了基因在不同萌发物候型茶树品种从秋季茶树顶芽开始停止生长到春季萌发期间以及茶树龙井43花芽发育阶段和种子萌发过程的表达量变化。如图7所示,在茶树芽休眠形成阶段,在茶树早生品种龙井43(除外)、中生品种碧云以及晚生品种政和大白芽中的表达量均达到最大值;在深休眠阶段,表达下调,并达到最低;而在萌发阶段,表达上调,但是仍然低于休眠形成阶段的水平。在萌发阶段,3个品种的表达量有所差异,可能是由于3个品种休眠解除时间的不同导致。在不同萌发物候期茶树品种芽中的表达量存在差异,并且龙井43>碧云>政和大白。其原因可能是此时龙井43已经萌发,生长较活跃;而碧云可能处于萌动阶段,政和大白则可能处于休眠后期。由此可见,茶树在芽停止生长开始到芽萌动阶段的表达模式与冬季芽休眠形成及解除高度关联。
茶树在母叶中的表达模式(图7-D—F)跟在芽中的有一定差别。在早生品种茶树龙井43叶片中,、有着相同的表达模式,即在休眠形成阶段,表达量处于较低水平,在萌芽阶段表达量达到最大,而是在深休眠阶段表达量最高,萌芽阶段表达量最低,则与相反,在深休眠阶段表达量达到最低,萌发阶段表达显著回升;在茶树中生品种碧云和晚生品种政和大白中,也在休眠形成阶段表达量最低。结果表明基因在不同萌发物候型茶树芽休眠形成和解除阶段过程中,在芽和母叶中的表达模式不同,对茶树的调控作用可能不同。
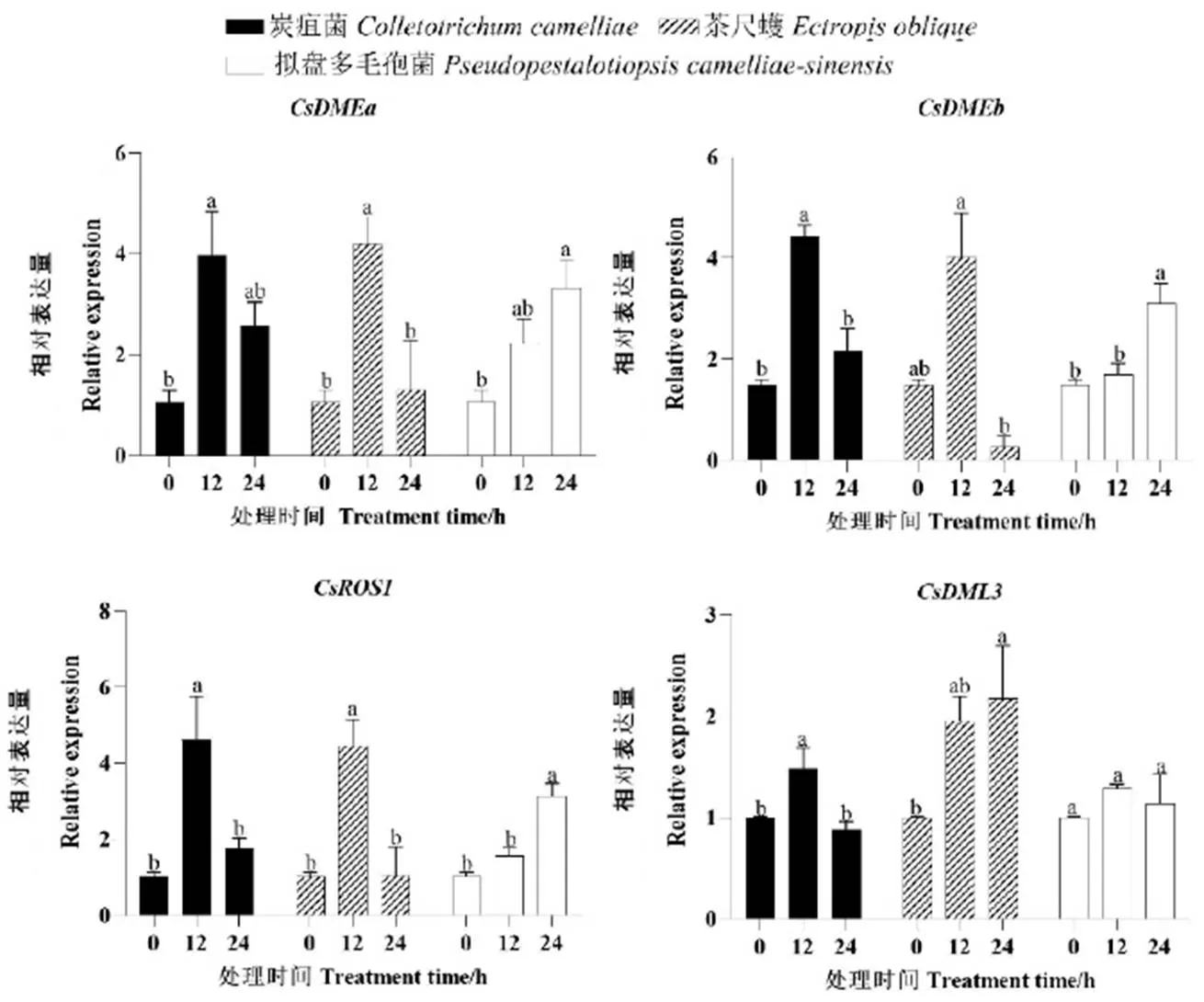
注:图中不同字母表示0.05水平上差异显著
利用qRT-PCR技术分析了基因在花芽发育阶段和种子萌发过程的表达量变化。由图8可以看出,在花芽发育阶段,在露白期表达显著上调,和在开放期显著下调。在花芽发育阶段的显著变化表明其在花的发育中起到调控作用。
注:图中不同字母表示0.05水平上差异显著
Note: Different letters in the figure indicate significant differences at the level of 0.05
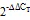
Fig. 6 Expression analysis ofunder drought stress in Longjing 43
注:(A)和(D)为茶树品种龙井43;(B)和(E)为茶树品种碧云;(C)和(F)为茶树品种政和大白。I:2018年10月25日(茶树休眠形成阶段);Ⅱ:2019年1月14日(茶树深休眠阶段);Ⅲ:2019年3月13日(茶树休眠解除阶段)。图中红色和蓝色分别表示较高和较低的表达水平(与白色相比)
Note: (A) and (D) Tea plant variety Longjing 43. (B) and (E) Tea plant variety Biyun. (C) and (F) Tea plant variety Zheng he da bai. I: October 25, 2018 (Paradormancy stage of tea plants). Ⅱ: January 14, 2019 (Endodormancy stage of tea plants). Ⅲ: March 13, 2019 (Encodormancy stage of tea plants). Compared with the white color, the red and blue colors in the graph indicate higher and lower expression levels, respectively
图7 茶树基因在不同茶树品种芽和叶休眠诱导下的表达分析
Fig. 7 Expression analysis ofin buds and leaves of different tea cultivars at different dormancy periods
在种子萌发前后也发生显著变化,在萌发种子中的表达量显著高于未萌发种子,约为萌发前的19倍;而在萌发种子中表达显著下调,表明和可能参与胚乳的表达调控,在种子萌发过程中起作用。
3 讨论
3.1 dMTases的蛋白质结构特征
DNA甲基化是影响许多生物学过程的重要表观遗传修饰,不仅参与基因表达的调节,而且还参与维持基因组的稳定性。dMTase可以去除甲基化并调节基因表达,与植物抗逆性密切相关,在表观遗传学中起着重要作用。目前,已在多种植物中进行了基因的鉴定,如拟南芥[21]、番茄[9]、蓖麻[22]、草莓[23]、花生[24]、棉花[25]等植物。本研究从茶树舒茶早中鉴定了4条基因,其具有已报道的植物的典型特征。系统进化分析将鉴定出的4个基因分成和两个分支,发现茶树的与葡萄和杨树的亲缘关系更密切;Gu等[21]将草莓中基因家族的4个基因归为两个分支,DME类基因存在于所有的双子叶植物中,在单子叶植物中不存在,且发现其在环境胁迫(盐、干旱、低温、高温)和草莓果实生长中具有调节作用。Cao等[9]将番茄中基因家族的3个基因归为2个分支,并发现其参与果实成熟的调控。基因结构及保守结构域分析表明,茶树基因家族聚类关系较近的基因具有相似的基因结构,如分支的和含有相同外显子数目。同时保守结构域的个数和种类具有一定相似性,在氮端都含有ENDO3c结构域,在碳端都含有DME结构域,这与葡萄、水稻、杨树的dMTase蛋白保守结构完全一致(图1)。说明这两个结构域是dMTase高度保守的结构域,编码由这两个结构域组成的蛋白质的基因可以被鉴定为推定的基因。
为了确定茶树中特定表观遗传基因可调控转录的因素,分析了它们在基因上游2 000 bp内推定的顺式作用元件的启动子序列。结果发现,的启动子大致具有光响应、植物激素响应、胁迫响应和生长发育4类顺式作用调控元件,其中胁迫响应类作用元件主要涉及低温应激、干旱诱导以及防御和应激反应,而生长发育相关的作用原件主要与种子特异性调控和胚乳特异性表达相关。
3.2 CsdMTase基因的表达与逆境胁迫的关系
在拟南芥中,转座因子序列(Transposable element sequences,TE)的DNA去甲基化(主要由ROS1完成)可以促进防御相关基因的表达,介导的启动子转座因子的DNA去甲基化对于防御相关基因表达的激活至关重要,并且dMTase调控的镰刀菌胁迫应答相关的基因表达具有组织特异性[28];茶树在接种炭疽菌、茶尺蠖和拟盘多毛孢菌处理的短时间内,基因的表达水平发生显著变化,基因相对表达量迅速增大,表明受生物胁迫的显著诱导,茶树通过DNA甲基化机制快速响应生物胁迫。目前,植物中dMTase对生物胁迫调节作用的相关研究还很缺乏,该结果可为日后dMTase对生物胁迫的调控作用提供依据。此外,在各种非生物胁迫下,基因的表达水平也发生了显著变化,这表明某些关键基因的去甲基化可能对植物中的非生物反应至关重要。油菜中,和在盐胁迫和高温胁迫下均温和上调,,和表现出对盐胁迫的一些下调响应,而在热胁迫下较少表达[27];蓖麻在干旱胁迫下基因显著上调;茶树在干旱胁迫时基因的表达水平也显著升高,尤其是基因的表达水平最高[13]。
注:(A)CsdMTases在茶树花芽发育过程的表达分析,S:幼蕾期,M:露白期,L:开放期。(B)CsdMTases在茶树种子萌发过程的表达分析,I:成熟种子,Ⅱ:萌发种子。图中不同小写字母表示0.05水平上差异显著;*表示P<0.05;**表示P<0.01
3.3 CsdMTase基因的表达与生长发育的关系
研究显示,在拟南芥中,幼嫩和成年拟南芥组织中的,和基因表达量增加[21];在杨树中,过表达会促进杨树的顶芽提前形成[11],而的沉默会导致杨树的芽萌发推迟[28],说明DNA去甲基化作用可调控芽的生长发育;在蓖麻中,在种子发育阶段显著上调[22],表明其可能在种子发育中具有潜在作用。在茶树中,基因在茶树种子萌发、花芽发育阶段显著上调,表明茶树可能在胚乳形成中起调节作用,并且通过参与的DNA去甲基化作用调控茶树的生长发育。在茶树休眠形成及解除阶段,基因在不同品种以及叶、芽中表达水平差异也很大,在茶树早生品种、中生品种、晚生品种中生理休眠期显著下调,而在生态休眠期表达量又有所上升,但是表达量依旧没有类休眠期的高,表明基因可能在茶树芽休眠的形成和解除过程中发挥着重要的作用。
DNA甲基化对于调节植物的发育和对环境胁迫应答至关重要,但是DNA甲基化酶和dMTase如何参与各种反应是一个复杂的过程,其机制仍不清楚。基因的差异表达分析表明,在不同逆境胁迫下,基因的表达水平发生了很大变化,一些关键基因可能被去甲基化。因此,DNA甲基化最有可能参与环境对茶树生长发育的影响。本研究为茶树中DNA甲基化对胁迫响应及其在种子萌发和花芽发育中的调控作用提供了一些线索,并为进一步探索茶树逆境胁迫以及生长发育过程中的表观遗传调控机制提供了基础。
[1] Law J A, Jacobsen S E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals [J]. Nat Rev Genet, 2010, 11(3): 204-220.
[2] Cao X, Jacobsen S E. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes [J]. PNAS 2002, 99(s4): 16491-16498.
[3] Duan C G, Zhu J K, Cao X. Retrospective and perspective of plant epigenetics in China [J]. J Genet Genomics, 2018, 45(11): 621-638.
[4] Rocha P S, Sheikh M, Melchiorre R, et al. Thehomology-dependent genegene codes for an S-adenosyl-L-homocysteine hydrolase required for DNA methylation-dependent gene silencing [J]. Plant Cell, 2005, 17(2): 404-417.
[5] Jon P, Huh J H, Tracy Ballinger, et al. DNA demethylation in thegenome [J]. PNAS 2007, 104(16): 6752-6757.
[6] Zhang H, Lang Z, Zhu J K. Dynamics and function of DNA methylation in plants [J]. Nat Rev Mol Cell Bio, 2018, 19(8): 489-506.
[7] Yeonhee Choi M G. Demeter, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in[J]. Cell Press, 2002, 110(1): 33-42.
[8] Gong Z Z, Teresa M R, Rafael R A, et al. ROS1, a repressor of transcriptional gene silencing in, encodes a DNA glycosylase/lyase [J]. Cell Press, 2002, 111(6): 803-814.
[9] Cao D, Ju Z, Gao C, et al. Genome-wide identification of cytosine-5 DNA methyltransferases and demethylases in[J]. Gene, 2014, 550(2): 230-237.
[10] Zemach A, Kim M Y, Silya P, et al. Local DNA hypomethylation activates genes in rice endosperm [J]. PNAS 2010, 107(43): 18729-18734.
[11] Conde D, Moreno C A, Dervinis C, et al. Overexpression of Demeter, a DNA demethylase, promotes early apical bud maturation in poplar [J]. Plant Cell and Environment Plant Cell Environ, 2017, 40(11): 2806-2819.
[12] 周艳华, 曹红利, 岳川, 等. 冷驯化不同阶段茶树DNA甲基化模式的变化[J]. 作物学报, 2015, 41(7): 1047-1055. Zhou Y H, Cao H L, Yue C, et al. Changes of DNA methylation levels and patterns in tea plant () during cold acclimation [J]. Acta Agronomica Sinica, 2015, 41(7): 1047-1055.
[13] Zhu C, Zhang S, Zhou C, et al. Genome-wide investigation and transcriptional analysis of cytosine-5 DNA methyltransferase and DNA demethylase gene families in tea plant () under abiotic stress and withering processing [J]. Peer J, 2020, 8: e8432. doi: 10.3389/fpls.2016.00007.
[14] 熊飞, 卢秦华, 房婉萍, 等. 基于全基因组的茶树PAL家族基因鉴定及其在生物与非生物胁迫下的表达分析[J]. 园艺学报, 2020, 47(3): 517-528. Xiong F, Lu Q H, Fang W P, et al. Genome-wide identification and expression analyses ofgenes under biotic and abiotic stress in[J]. J Hortic, 2020, 47(3): 517-528.
[15] Xia E, Tong W, Hou Y, et al. The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into genome evolution and adaptation of tea plants [J]. Mol Plant, 2020, 13(7): 1013-1026.
[16] Wei C, Yang H, Wang S, et al. Draft genome sequence ofvar.provides insights into the evolution of the tea genome and tea quality [J]. PNAS 2018, 115(18): E4151-E4158.
[17] Xia E H, Li F D, Tong W, et al. Tea plant information archive: a comprehensive genomics and bioinformatics platform for tea plant [J]. Plant Biotechnol J, 2019, 17(10): 1938-1953.
[18] Hao X, Horvath D P, Chao W S, et al. Identification and evaluation of reliable reference genes for quantitative real-time PCR analysis in tea plant ((L.) O. Kuntze) [J]. Int J Mol Sci, 2014, 15(12): 22155-22172.
[19] Thomas D. Analyzing real-time PCR data by the comparative CTmethod [J]. Protocol, 2008, 3(6): 1101. doi: 10.1038/nprot.2008.73.
[20] Mok Y G, Uzawa R, Lee J, et al. Domain structure of the DEMETER 5-methylcytosine DNA glycosylase [J]. PNAS 2010, 107(45): 19225-19230.
[21] Ogneva Z V, Dubrovina A S , Kiselev K V . Age-associated alterations in DNA methylation and expression of methyltransferase and demethylase genes in[J]. Biol Plantarum, 2016, 60(4): 628-634.
[22] Victoria D, Aliki K, Venetia K, et al. Spatial and temporal expression of cytosine-5 DNA methyltransferase and DNA demethylase gene families of theduring seed development and drought stress [J]. Plant Growth Regul, 2017, 84(1): 81-94.
[23] Gu T T, Ren S A, Wang Y H, et al. Characterization of DNA methyltransferase and demethylase genes in[J]. Mol Genet Genomics, 2016, 291(3): 1333-1345.
[24] Wang P F, Chao G, Bai X T, et al. Genome-wide identification and comparative analysis of cytosine-5 DNA methyltransferases and demethylase families in wild and cultivated peanut [J]. Front Plant Sci, 2016, 7: 7. doi: 10.3389/fpls.2016.00007.
[25] Yang X M, Chen X G, Wang D L. Genome-wide identification and expression analysis of DNA demethylase family in cotton [J]. J Cotton Res, 2019, 2(2): 142-150.
[26] Ulrike S, Joanne L, Kemal K, et al. DNA-demethylase regulated genes show methylation-independent spatiotemporal expression patterns [J]. Front Plant Sci, 2017, 8: 1449. doi: 10.3389/fpls.2017.01449.
[27] Fan S, Liu H, Liu J, et al. Systematic analysis of the DNA methylase and demethylase gene families in rapeseed (.) and their expression variations after salt and heat stresses [J]. Int J Mol Sci, 2020, 21(3): 953. doi: 10.3390/ijms21030953.
[28] Conde D, Le G A, Perales M, et al. Chilling-responsive Demeter-Like DNA demethylase mediates in poplar bud break [J]. Plant Cell Environ, 2017, 40(10): 2236-2249.
Genome-wide Investigation and Expression Analysis of DNA Demethylase Genes in Tea Plant ()
CHEN Yao, ZHANG Weifu, REN Hengze, XIONG Fei, ZHANG Haojie, YAO Lina, LIU ying, WANG Lu, WANG Xinchao, YANG Yajun*, HAO Xinyuan*
Tea Research Institute of the Chinese Agriculture Science/National Center for Tea Improvement/Key Laboratory of Tea Biology and Resources Utilization, Ministry of Agriculture and Rural Affairs, Hangzhou 310008, China
DNA demethylase (dMTase) is a bifunctional enzyme with activities of glycosylase and apyrimidic lysase during active demethylation. Based on the available genome data of tea plant, bioinformatics analysis was used to comprehensively identify DNA demethylases () in tea plant. The genome-wide identification results show that the tea cultivar Shuchazao contains 4. Thesecould be divided into two branches (and) with differences in gene length and intron number by phylogenetic and gene structure analysis. Further analysis shows that the physical and chemical properties of different CsdMTase proteins are similar, which contain the typical conserved domains of ENCO3c and RRM_DME and are located in the nucleus. The promoter regions ofcontain a large number of-acting components related to light response, plant hormone response, stress response and growth and development. Thegenes are expressed in all detected tissues with certain tissue specificity. The expressions ofwere significantly up-regulated under biotic stresses, including,andtreatments. During winter dormancy, different expression patterns ofwere detected in mature leaves and overwintering buds of different cultivars. The expressions ofandat different stages of flower bud development and seed germination were significantly induced. The results show that active DNA demethylation process mediated byplays a crucial role in regulating the stress responses, growth and development of tea plant, providing a theoretical basis for discovering the epigenetic regulation mechanism in tea plant.
tea plant, DNA demethylase, stress response, growth and development, expression analysis
S571.1;Q523
A
1000-369X(2021)01-028-12
2020-05-27
2020-06-17
国家自然科学基金(31972461)
陈瑶,女,硕士研究生,主要从事茶树遗传育种研究。*通信作者:yjyang@tricaas.com,haoxy@tricaas.com
(责任编辑:赵锋)
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
上海师范大学学报·自然科学版(2022年3期)2022-07-11
中国农学通报(2022年13期)2022-05-31
茶道(2022年3期)2022-04-27
昆明医科大学学报(2022年1期)2022-02-28
农学学报(2021年11期)2021-12-12
云南画报(2021年10期)2021-11-24
实用肿瘤学杂志(2020年4期)2020-12-08
延河·绿色文学(2016年8期)2016-05-14
小学生导刊(中年级)(2014年4期)2014-05-09
