
一步法构建莱茵衣藻叶绿体表达载体
2021-03-04 07:40刘国兴胡玉立徐丹丹
现代农业研究 2021年3期
刘国兴,胡玉立,徐丹丹,徐 松
(国药集团动物保健股份有限公司 湖北,武汉 430075)
莱茵衣藻是一种生活在淡水里的单细胞真核绿藻,其叶绿体表达的外源蛋白具有安全性好、培养周期短、遗传稳定、容易扩大培养,并且成本低廉等优点[1,2,3]。近些年来,多种具有应用前景的蛋白在莱茵衣藻叶绿体中成功表达[4]。而一般构建衣藻叶绿体表达载体需要通过两步克隆来完成:首先将外源基因插入克隆载体的衣藻来源的5’和3’非翻译区(UTR)多克隆位点间,再将5’UTR-外源基因-3’UTR的表达盒酶切下,通过同源重组构建至表达载体[5,6]。为了简化衣藻叶绿体表达载体的构建步骤,本研究构建了一种T载体pBTNK,通过一步TA克隆可将待表达的外源基因插入衣藻来源5’和3’UTR 之间,蓝白斑筛选获得阳性转化子后,重组质粒可直接用于转化植物,因此可简化衣藻叶绿体中表达载体构建流程。
1 材料和方法
1.1 材料
1.1.1 藻株、菌株和质粒 莱茵衣藻Chlamydomonas reinhardtii137c (mt)、质粒 pCX16、pCX13、p228 由华中农业大学陈杏洲博士惠赠;大肠杆菌DH10β,质粒pShuttle、pFAST-EGFP、pT-BASIC由湖北大学蒋思婧博士惠赠。
1.1.2 酶、主要试剂和设备 BfuI 购自Thermo Scientific,LATaq酶、SolutionI 连接酶、T4DNA 聚合酶、质粒抽提试剂盒、MiniBEST Agarose Gel DNA Extraction Kit Ver.4.0、其他限制性内切酶,购自 Takara 公司,X-gal、dNTP、硫酸卡那霉素(kana)、氨苄青霉素购自北京九州同业生物科技公司,壮观霉素购自sigma公司,PCR引物(表1)由上海捷瑞生物工程有限公司合成,Biolistic PDS-1000/He基因枪购于Bio-Rad。
1.2 方法
1.2.1 DNA 克隆 质粒提取参照OMEGA bio-tek 试剂盒说明书进行,DNA片段克隆、重组质粒鉴定等分子克隆操作参照Sambrook提供的方法进行[7]。
1.2.2 莱茵衣藻叶绿体转化 根据文献[8]描述的方法并适当调整后,通过基因枪法将基因导入叶绿体,即3μg载体质粒pTBNK-EGFP(1μg/μl)和2μg 抗性质粒p228(1 μg/μl)包被50μl(60 mg/mL)直径为0.6μm金粉子弹,按压力1100psi、真空度28英寸汞柱、目标距离6cm轰击衣藻,将轰击后的衣藻在TAP平板上暗培养15~20h后刮下,涂布于5皿含100μg/mL壮观霉素抗性TAP平板,培养两周。
1.2.3 叶绿体总DNA提取 衣藻叶绿体总DNA的抽提采用CTAB法[9]。
1.2.4 莱茵衣藻的培养 将莱茵衣藻培养在TAP 液体培养基或含1%琼脂糖的固体平板上,在22℃~24℃、连续光照(约100 μE/m2.sec-1)条件下培养[10]。
2 结果
2.1 衣藻叶绿体表达载体pTBNK的构建
以质粒pCX16为模板,P1和P2为引物,LATaq酶PCR扩增5.7kb 同源臂上的2.0 kb 片段,回收PCR 产物后,用BamHI和SphI双酶切并回收该2.0 kb片段,然后将酶切的片段插入pCX16 载体的BamHI 和SphI 酶切位点间,在pCX16 载体上引入NotI 酶切位点得到载体pCXN;EcoRI和SphI 双酶切载体pCXN,回收pCXN 载体骨架5.7 kb 片段,并用T4DNA 聚合酶补平末端,将末端补平片段插入pT-BASIC 的EcoRV 酶切位点得到载体pTBN;以质粒pShuttle 为模板、P3 和 P4 为引物,LATaq酶 PCR 扩增 1.2 kb kana 抗性基因片段,将该片段回收后,BglⅡ和HindⅢ双酶切该片段,然后将酶切后的片段插入pCX13 载体的BglⅡ和HindⅢ酶切位点间获得载体pCX13-kana;用BamHI 将 pCX13-kana 载体上 5’UTR-BfuI-kana-BfuILacO 部分序列-3’UTR 表达盒酶切下,T4 DNA 聚合酶补平末端,然后和NotI 酶切、T4DNA 聚合酶补平末端的pTBN 载体片段用SolutionI 连接酶进行连结,获得衣藻叶绿体定向表达T载体pTBNK(图1)。
2.2 一步克隆构建衣藻叶绿体表达载体pTBNK-EGFP
以 pFAST-EGFP 为模板,P5 和 P6 为引物,LAtaqPCR扩增 EGFP 基因并回收该产物,BfuI 酶切 pTBNK 载体,连接后转化至大肠杆菌DH10β 感受态细胞,涂布于含氨苄青霉素(50μg/ml)和X-gal(30μg/ml)的LB固体平板上,避光培养12~14h 后观察,挑显蓝色的菌落(图2)进行PCR鉴定(图3)。经鉴定,阳性克隆达100%,表明重组衣藻表达载体pTBNK-EGFP构建成功。
2.3 表达载体pTBNK-EGFP导入衣藻叶绿体
pTBNK-EGFP 经基因枪法转化莱茵衣藻叶绿体后,经100μg/mL壮观霉素抗性筛选,共长出4个绿色单藻落,抽提野生型的叶绿体总DNA和第八次传代的转基因衣藻的叶绿体总 DNA 作为模板,以 P5 和 P6 为引物,PCR 扩增EGFP片段,结果显示四个转基因衣藻DNA样本中均可以扩增出目的条带而野生型DNA样本无法成功扩增目的条带(图4),表明EGFP成功插入莱茵衣藻叶绿体。
3 讨论
我们设计在载体克隆位点引入BfuI 酶切位点,该酶的特点是特异性识别5’-gtatcc-3’序列,并在该序列下游任意6 个碱基处对DNA 进行切割,切割后形成粘性末端“T”,而LAtaqDNA 聚合酶扩增的目的片段带“A”尾。这样,可通过TA克隆将目的片段克隆到pTBNK。
基于LacO重构原理[11],在PCR 扩增目的片段的下游引物5’增加7bp的部分LacO序列(5’-CACAATT-3’),载体骨架上携带 13bp 的部分LacO序列(3’-AATTGTGAGCGCT-5’),载体片段和目的片段连接后,转化到lac+的菌株感受态细胞,涂布于含X-gal的平板,完整的LacO序列会竞争性的吸附宿主菌表达的LacI 蛋白,使宿主菌的乳糖启动子控制的β-半乳糖苷酶基因表达解抑制,表达β-半乳糖苷酶,分解X-gal使菌落呈现蓝色。因此,只有目的基因阅读框正确插入克隆位点组成完整LacO序列才会使菌落显蓝色,而基因阅读框反向插入或空载体,菌落均呈白色。统计显示,EGFP基因克隆至该载体并转化后,蓝色菌落占总菌落数的50%~60%,而且蓝色菌落均为阅读框正确插入的重组子(图3)。
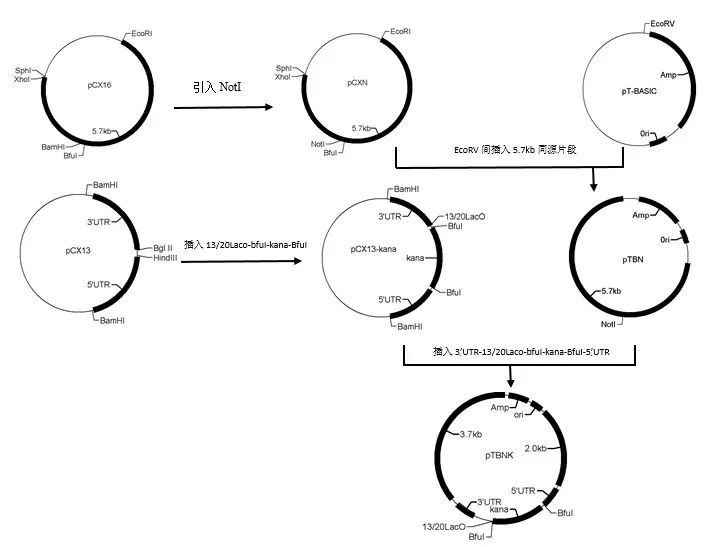
图1 衣藻叶绿体定向表达T载体构建流程图
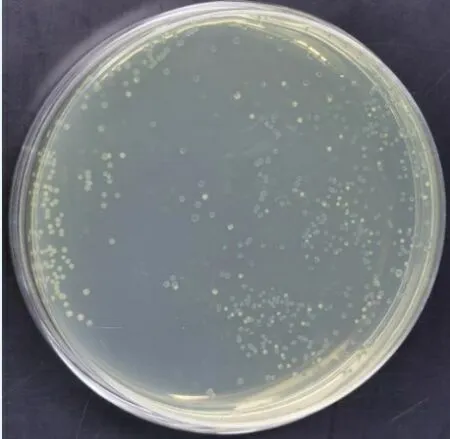
图2 蓝白斑筛选重组子平板

图3 1:DNAmmaakkeerr 2Kpluuss IIII;2~2200:蓝斑菌落PPCCRR扩增EEGGFFPP基因;2211~2255:白斑菌落PPCCRR扩增
本实验构建衣藻叶绿体表达载体pTBNK,克隆效率高、筛选方法简便、耗时短、成本低,而且不受酶切位点的影响,具备通用性,且重组子可通过蓝白班筛选,优于传统的两步法酶切构建表达载体[7,11]。遗憾的是,本实验中EGFP未表达。尽管如此,该构建方法极大简化外源基因插入莱茵衣藻叶绿体表达载体的步骤,载体构建思路也可能适用于其他叶绿体表达载体的构建。

图4 1:DNAmmaakkeerr 2Kpluuss IIII;2~5:转基因衣藻抽提总DDNNAA作为模板PPCCRR 扩增EEGGFFPP 基因;6:野生型衣藻抽提总DDNNAA 作为模板PPCCRR扩增EEGGFFPP基因
猜你喜欢
军事文摘(2022年21期)2022-11-17
现代仪器与医疗(2021年2期)2021-07-21
安徽农业科学(2021年4期)2021-03-18
中国当代医药(2020年26期)2020-11-06
生物信息学(2020年2期)2020-07-09
深圳大学学报(理工版)(2018年5期)2018-09-26
安徽农业科学(2018年18期)2018-05-14
分析化学(2015年10期)2015-11-03
