
芴类衍生化合物光电性能的密度泛函模拟计算
2021-03-13 14:38翁闻升肖慧萍曹家庆
现代信息科技 2021年15期
翁闻升 肖慧萍 曹家庆




摘 要:芴结构的化合物广泛被用于有机光电领域。光电性质的理论模拟计算对设计合成性能卓越的有机光电材料具有一定指导作用。根据密度泛函理论,采用高斯软件针对芴类衍生化合物的偶极矩、HOMO与LUMO的具体分布情况及吸收光谱等光电性能进行了模拟计算,研究了氧杂环丁烷基团、砜基和胺基的引入对芴类衍生物光电性能的影响。结果表明:氧杂环丁烷基团对芴类分子的光电性能几乎没有影响;砜基和胺基的引入能分别降低芴类衍生物的LUMO能级与HOMO能级;含砜基和胺基的芴类衍生物偶极矩较大,极性溶剂可作为其良溶剂。
关键词:芴类衍生化合物;高斯软件;光电性能;氧杂环丁烷基团;砜基;胺基
中图分类号:TP391.9;O626 文献标识码:A文章编号:2096-4706(2021)15-0145-04
Abstract: Compounds with fluorene structure are widely used in the field of organic optoelectronics. Theoretical simulation calculation of photoelectric properties has a certain guiding role in the design and synthesis of organic photoelectric materials with excellent performance. Based on the density functional theory, Gaussian software is used to simulate the optoelectronic properties of fluorene derivative compounds, such as the dipole moments, the specific distribution of HOMOs and LUMOs, and the absorption spectrum, exploring influence of sulfone group, oxetane group and amine group on the photoelectric properties of fluorene derivatives. The results show that the oxetane side chain group has almost no effect on the optoelectronic performances of fluorene molecules; the introduction of sulfone group and amine group can effectively reduce the LUMO energy level and the HOMO energy level of fluorene derivatives, respectively; fluorene-based materials containing sulfone group and amin units have large dipole moments and polar solvents can be used as their good solvents.
Keywords: fluorene derivatives; Gaussian software; optoelectronic performances; oxetane group; sulfone group; amine group
0 引 言
在科學技术飞速进步的今天,对材料性能的理论模拟计算已然是研究者们一个不可或缺的辅助研究工具。其中,密度泛函理论(DFT)是有效的计算方式之一。为了提高有机光电器件的性能,寻找更加适用的新型光电材料,研究者们采用DFT成功预测和计算了一系列共轭化合物及聚合物的能隙和光谱,并指导于实验,与实验值对比,取得相似的结果[1-5],这说明DFT计算结果对实验的方向具有一定指导作用。
芴结构的化合物,具有特殊的刚性稠环结构;并且芴类衍生物的结构很容易进行修饰,所以可以方便地在芴环上引入各种功能基团[5]。芴类衍生物已被广泛用于有机光电领域[6,7]。砜基是一种电负性较强的基团,因此具有良好的电子传输和电子注入能力。将其引入到化合物中,能够有效地修改最高占据分子轨道和最低未占据分子轨道(HOMO和LUMO),以提高光电性能[8-10]。氧杂环丁烷基团属于含能杂环,在芴的化学结构中引入氧杂环丁烷基团,可以制得较好的含能衍生物[11-14]。胺基能赋予化合物良好的水/醇溶性,加工性能更加环保;同时,也可以与其他使用有机溶剂进行加工的材料交叉形成叠层膜,而不会导致溶剂互溶,最近也得到较多关注[6-11]。
基于此,为了分析氧杂环丁烷基团、砜基和胺基的引入对芴类衍生化合物的光电性能是否有影响,本文根据密度泛函理论,利用高斯软件模拟计算了分别含有氧杂环丁烷基团、砜基和胺基侧链的芴类衍生化合物的光电性能。而目前对于这类芴类衍生化合物光电性能的高斯模拟计算未见报道。
1 计算模型和模拟计算方法
本文设计了如图1所示化学结构的化合物分子模型,其中,模型2、3和4分别为含有氧杂环丁烷基团、胺基和砜基的芴分子。另外,模型1(烷基芴分子)作为对比。
使用ChemOffice软件的ChemDraw功能绘制化合物的结构式。根据密度泛函理论,利用Gaussian 09程序包对化合物的光电性能进行计算[15],具体为:在6-31G(dp)基组下采用B3LYP对分子的基态结构优化,在此基础上再采用含时密度泛函(TD-DFT)模拟计算芴类衍生物的HOMO/LUMO能级及其能级差△H-L和最小激发能(Eg)。
2 光电性能的高斯模拟计算
2.1 分子结构的优化
为了保证计算的流畅性,使用Chem3D对所绘分子结构的构型进行进一步优化。将初步优化后的结构导入Gaussian中用DFT进行基态结构全优化,得到具有最低能量的分子构型,如图2所示。
2.2 偶极矩的模拟计算结果
一般来说分子的极性是由分子的偶极矩决定的,而根据极性相近原则,分子的极性越大,越容易溶解于强极性溶剂。对于有机光电材料来说,其优势在于可以通过溶液加工法大量运用于实际中的器件制备,因此需要有良好的溶解加工性能。化合物1、2、3和4的偶极矩模拟计算结果列于表1。
由表1可见,相对于化合物1,化合物2、3、4的总偶极矩更高,说明氧杂环丁烷基团、胺基和砜基等基团的引入能够提高芴类化合物的偶極矩。X、Y、Z轴任一轴上偶极矩的变化都影响到总偶极矩的变化。对比化合物1与2,化合物2相比化合物1在X和Z轴上的偶极矩变化很明显,导致化合物2有较大的总偶极矩,说明氧杂环丁烷基团的引入可增大分子的偶极矩,因此,化合物2相对化合物1而言在极性溶剂中具有更好的溶解性。化合物4在Y轴上的偶极矩比其他三种化合物高许多,导致其总偶极矩也明显增大,即在芴分子单元上引入砜基比含胺基和氧杂环丁烷基团的化合物的偶极矩都要大,相应表现出更大的极性。由此可见,化合物2、4在极性溶剂中的溶解性能相对于化合物1、3更大,即在极性溶剂中的溶解性排序:4>2>3>1。
2.3 前线分子轨道的模拟计算结果
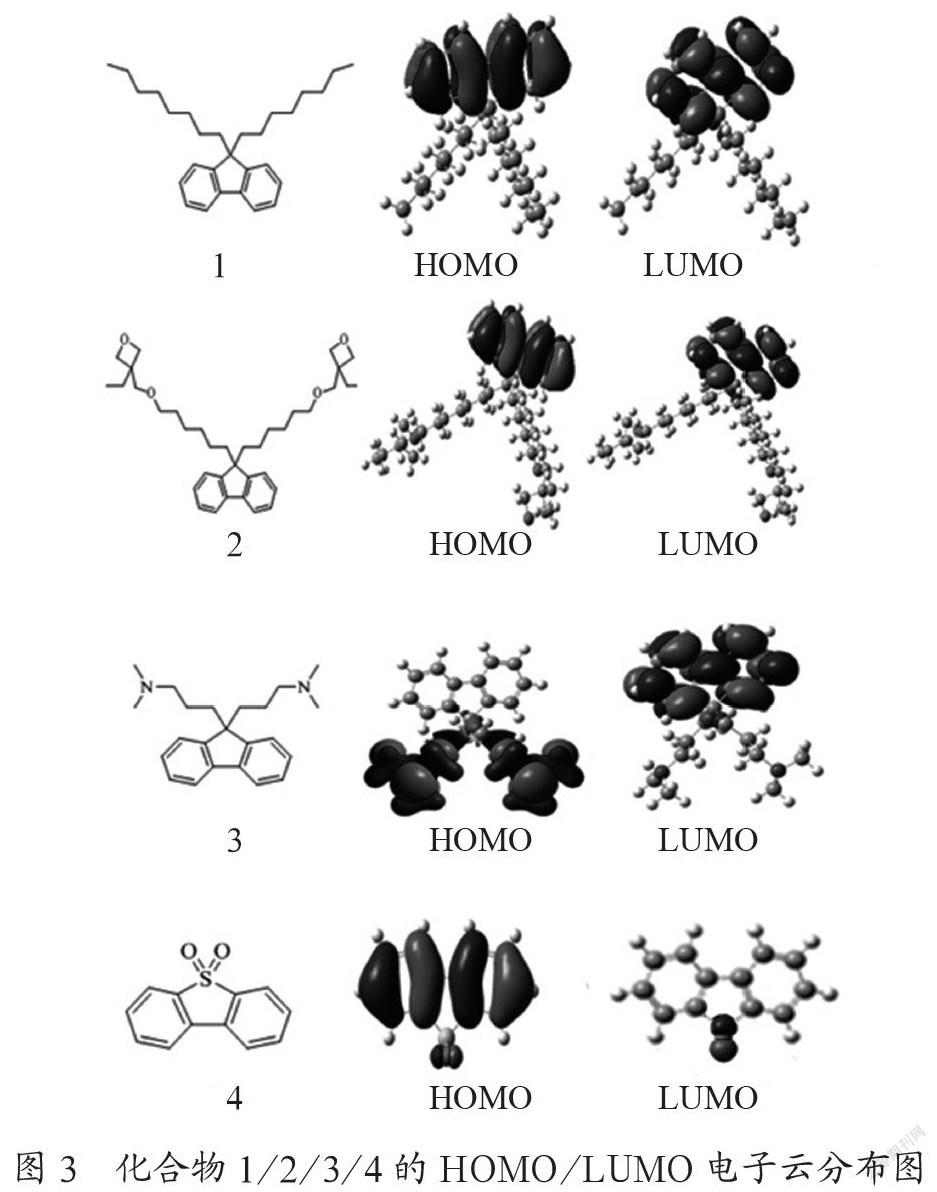
前线轨道理论认为,对一个分子的性质起决定性作用的是其HOMO和LUMO能级[16]。化合物1、2、3与4的HOMO/LUMO分布如图3所示。
从图3可知,化合物1与2的HOMO、LUMO分布相似,均匀分布于芴的共轭苯环上,侧链上没有电子云分布,说明氧杂环丁烷侧链对芴环的电子云分布情况没有影响。而与化合物1相比,化合物3、4的HOMO、LUMO分布明显不同:化合物3的HOMO和LUMO分别分布于胺基侧链上和芴的共轭苯环上,这说明胺基具有给电子性;化合物4的HOMO和LUMO分别分布于共轭苯环和砜基上,这说明砜基具有吸电子能力。由此可知:氧杂环丁烷侧链对芴环的电子云分布情况几乎没有影响;而胺基、砜基侧链对芴环主体结构的电子云分布情况有一定的影响。
HOMO和LUMO轨道的能量差构成了材料的能带隙,但是由于实验条件的限制,一般很难精确测得,通常采用最大光谱吸收的能量或基于循环伏安法起峰时的能量来衡量,因此,HOMO-LUMO(△H-L)值往往与实际情况不太相符。为了准确预测化合物的能级变化趋势,采取高斯软件中的TD-DFT方法来计算最低激发能Eg(即:紫外光谱最大吸收的能量)。虽然两者之间的数值有些差别,但是趋势是一致的,一般为了简化计算可以用△H-L来进行预测。模拟计算的结果列于表2。
由表2可知:化合物1和和2的轨道能级、带隙和最低激发能几乎是一致的,说明氧杂环丁烷侧链的引入对芴基化合物的能级影响不大。化合物3的HOMO能级明显高于化合物1、2和4的HOMO能级,说明胺基具有给电子性。化合物4的HOMO/LUMO能级相对化合物1的-5.83 eV和-0.85 eV变得更深,这种能级上的变化进一步证实了砜基的缺电子性;相对化合物1、2而言,化合物3、4的△H-L和Eg均较小,也就是说,从HOMO能级到LUMO能级所需能量更少,电子跃迁更容易发生。
2.4 分子的吸收光谱分析
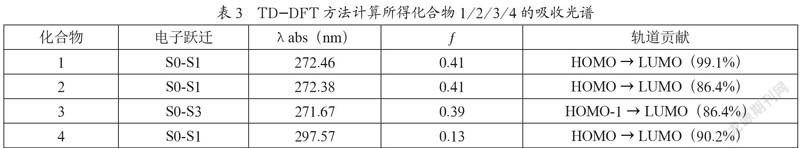
在优化的基态分子结构上采用TD-DFT进一步计算了化合物1、2、3和4的吸收光谱,分析其中振子最强时的最大吸收波长,具体见表3。
从表3中的电子迁跃信息可以得出,化合物1、2、4电子跃迁主要发生在S0-S1且主要贡献来自于HOMO→LUMO。化合物3的S0-S1所对应的最大波长处振子强度(f)很低,小于0.01,基本上不发生迁跃,而电子跃迁主要发生在S0-S3,HOMO-1能级与HOMO能级有着类似的电子分布,电子主要从胺基的孤电子对向芴环跃迁。由最大吸收波长可以看出,氧杂环丁烷基团和胺基基团对吸收波长的影响很小,而砜基对吸收波长的影响较大。
3 结 论
通过论证分析总结,氧杂环丁烷侧链基团的引入对芴类分子的光电性能几乎没有影响。砜基和胺基的引入能分别降低芴类衍生物的LUMO能级与HOMO能级。含砜基和胺基的芴类衍生物偶极矩较大,极性溶剂可作为其良溶剂。
参考文献:
[1] 李高楠,王鹏江,夏东,等.新型 9-丙二腈芴类衍生物的合成及光谱性质的理论研究[J].分子科学学报,2015,31(3):245-250.
[2] 韩立志,王卓,华英杰,等.9,9-二-(3-(9-苯咔唑基))-2,7-芘基芴的光电性质 [J].化学学报,2012,70(5):579-584.
[3] FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al Gaussian 09,Revision E. 01 [EB/OL].[2021-04-26].https://www.scienceopen.com/book?vid=f349ad30-c1e1-4d1d-960e-120276f311d4.
[4] VANASUNDARI K,BALACHANDRAN V,KAVIMANI M,et al. Spectroscopic investigation,vibrational assignments,Fukui functions,HOMO-LUMO,MEP and molecular docking evaluation of 4-[(3,4-dichlorophenyl) amino] 2-methylidene 4-oxo butanoic acid by DFT method [J].Journal of Molecular Structure,2017,1147:136-147.
[5] SHEN Y P,CAI H X,CHEN F Y,et al. A relativistic DFT probe for small-molecule activation mediated by low-valent uranium metallocenes [J].New Journal of Chemistry,2021,45(9):4270-4279.
[6] HUANG F,NIU Y H,ZHANG Y,et al. A conjugated,neutral surfactant as electron-injection material for high-efficiency polymer light-emitting diodes [J].Advanced Materials,2007,19(15):2010-2014.
[7] HOU Q,XU Y,YANG W,et a1. Novel Red-emitting Fluorene Based Copolymers [J].Journal of Material Chemistry,2002,12(10):2887-2892.
[8] CHAN K L,WATKINS S E,MAK C S K,et al. Poly(9,9-dialkyl-3,6-dibenzosilole)--a high energy gap host for phosphorescent light emitting devices [J].Chemical Communications,2005(46):5766-5768.
[9] XIAO H,CAO J Q,ZHOU J P,et al. Study on the synthesis and optoelectronic properties of the homopolymer of 2,8-bis(octyloxy)-dibenzothiophene-S,S-dioxide [J].Synthetic Metals,2020,260:116285.
[10] HUANG C Y,CHENG C Y,SHIH Y S. All-solution-processed fluorene/dibenzothiophene-S,S-dioxide blue co-oligomer light-emitting diodes with an electron transporting PEI/ultrafine-ZnO-nanoparticle bilayer [J].RSC advances,2017,7(66):41855-41861.
[11] ZHANG K,ZHONG C M,LIU S J,et al. Highly efficient inverted polymer solar cells based on a cross-linkable water-/alcohol-soluble conjugated polymer interlayer [J].ACS Applied Materials and Interfaces,2014,6(13):10429-10435.
[12] YAN Y,LI W,CAI J L,et al. Improved efficiency in fullerene and non-fullerene polymer solar cells having an interdigitated interface with the electron transport layer [J].Materials Chemistry Frontiers,2018,2(10):1859-1865.
[13] SONG C,HU Z H,LUO Y,et al. Organic/inorganic hybrid EIL for all-solution-processed OLEDs [J].Advanced Electronic Materials,2018,4(2):1700380.
[14] HSU F C,LIN J Y. Effects of thermally cross-linkable polymeric additive in the photoactive layer of polymer solar cells [J].Organic Electronics,2019,67:128-135.
[15] MEINEL B,KOSCHWITZ T,BLOCKS C,et al. Comparison of diamond wire cut and silicon carbide slurry processed silicon wafer surfaces after acidic texturisation [J].Materials Science in Semiconductor Processing,2014,26:93-100.
[16] VANASUNDARI K,BALACHANDRAN V,KAVIMANI M,et al. Spectroscopic investigation,vibrational assignments,Fukui functions,HOMO-LUMO,MEP and molecular docking evaluation of 4-[(3,4-dichlorophenyl) amino] 2-methylidene 4-oxo butanoic acid by DFT method [J].Journal of Molecular Structure,2017,1147:136-147.
作者簡介:翁闻升(1996—),男,汉族,江西南昌人,硕士研究生在读,主要研究方向:计算机在材料中的应用;肖慧萍(1977—),女,汉族,副教授,硕士生导师,博士,主要研究方向:计算机在材料中的应用。
3957500338263
