
KRN7000的合成研究
2021-03-25 09:51郝玉伟邢露匀曹梦涵毕晶晶
河南化工 2021年2期
郝玉伟,邢露匀,曹梦涵,毕晶晶
(河南师范大学 化学化工学院,河南 新乡 453007)
随着对肿瘤免疫认识和T细胞识别机制的突破性研究进展,神经鞘糖脂在肿瘤、肿瘤转移的诊治和预防中开辟了一条全新的免疫治疗途径[1-3]。1990年代初,日本麒麟啤酒有限公司从冲绳海绵Agelas Mauritianus的提取物中筛选出一系列鞘糖酯类化合物,并命名为Agelasphins (AGLs)[4]。它们是D-半乳糖以α-糖苷键与神经酰胺类化合物连接而成。其中α-半乳糖神经酰胺(α-GalCer,KRN7000)作为明星分子,受到广泛关注[5-7]。研究表明α-GalCer具有抗肿瘤及激活免疫作用,能通过激活NKT细胞参与多种自身免疫性疾病、肿瘤、动脉粥状硬化和感染性疾病的免疫调节中,它作为抗癌新药已进入Ⅰ期临床研究阶段,是药物学家关注的重要免疫调节剂[8-9]。
KRN7000通常是由半乳糖基供体和神经酰胺受体通过糖苷反应构建骨架,进而脱除各种保护基而成。所以对于KRN7000的合成来说,需要构建糖基化反应的供体和受体两个模块。近年来,对KRN7000的合成研究主要有如下几种方法。如TAKIKAWA等[10]通过1-氟-2,3,4,6-四-氧-苄基-α-D-半乳吡喃糖与2-(N-二十六烷酰基氨基)-3,4-二-O-叔丁基二甲基硅基-植物鞘氨醇在SnCl4/AgClO4的催化下以四氢呋喃(THF)为溶剂在-10 ℃下进行偶联反应(收率为53%)。KIM等[11]通过2,3,4,6-四-氧-苄基-α-D-半乳吡喃糖施密特试剂与2-(N-二十六烷酰基氨基)-3,4-二-O-叔丁基二甲基硅基-植物鞘氨醇在三氟化硼乙醚(BF3·Et2O)的催化下,以THF-Et2O为溶剂在-20 ℃下进行偶联反应(收率为63%)。通过2,3,4,6-四-氧-苄基-α-D-半乳吡喃糖施密特试剂与2-(N-二十六烷酰基氨基)-3,4-二-O-苯甲酰基-植物鞘氨醇在三氟甲磺酸三甲硅酯(TMSOTf)的催化下,以THF-Et2O为溶剂在-23 ℃下进行偶联反应(收率为59%)。DU等[12]通过1-碘代-2,3,4,6-四-氧-苄基-α-D-半乳吡喃糖与2-叠氮基-3,4-二-O-对甲氧基苄基-植物鞘氨醇在四丁基碘化铵-N,N-二异丙基乙胺(TBAI-DIEA)的作用下以苯为溶剂在65 ℃下加热进行偶联反应。
以上方法用到的糖基供体为氟代糖、碘代糖或者施密特试剂,它们的稳定性相对较差,合成步骤长且复杂,不适合工业大批量合成,保存条件相对来说更加苛刻[13-15]。硫苷糖的稳定性相对较高,不易变质,很容易大批量合成和保存。且前三种方法需要在低温条件下进行偶联反应,第四种方法所用的受体需要将植物鞘氨醇的氨基进行叠氮化,偶联后得到α-GalCer骨架后还需要在经过氨基化、酰胺化和脱保护才能得到目标化合物。总体路线长,收率偏低。因此本研究尝试了一种新的合成路线,以2,3,4,6-四-氧-苄基-α-D-半乳吡喃糖硫苷作为一种稳定易得的糖基供体,与2-(N-二十六烷酰基氨基)-3,4-二-O-苯甲酰基-植物鞘氨醇在N-碘代丁二酰亚胺(NIS)/TMSOTf的作用下以二氯甲烷为溶剂在0 ℃下进行偶联反应,进而在甲醇钠的作用下脱除苯甲酰基,在H2,Pd(OH)2/C的作用下脱除苄基,即可获得KRN7000,总共3步,总收率达57.3%。
1 实验部分
1.1 主要试剂和仪器
N-碘代丁二酰亚胺(北京百灵威科技有限公司),二氯甲烷、甲醇、石油醚、乙酸乙酯(天津市德恩化学试剂有限公司),0.4 nm分子筛、三甲基硅基三氟甲基磺酸酯、Pd(OH)2/C(北京伊诺凯科技有限公司),三乙胺(天津市天力化学试剂有限公司),Na2S2O3、甲醇钠(天津市天力化学试剂有限公司),酸性树脂IR-120(郑州阿尔法化工有限公司),以上药品均为分析纯。
R-1001VN旋转蒸发仪(郑州长城科工贸有限公司),核磁共振仪-Avance 400 MHz (瑞士布鲁克公司),Bruker micro ToF II高分辨率质谱(德国布鲁克公司),BSA224S电子天平(赛多利斯科学仪器有限公司),DHG-9140A型电热恒温鼓风干燥箱(郑州国瑞仪器有限公司),ZF-20D暗箱式紫外分析仪(巩义市予华仪器责任有限公司)。
1.2 实验方法
1.2.1KRN7000的合成原理
以2,3,4,6-四-氧-苄基-α-D-半乳吡喃糖硫苷与2-(N-二十六烷酰基氨基)-3,4-二-O-苯甲酰基-植物鞘氨醇为起始原料,首先在NIS/TMSOTf的作用下通过糖基化反应得到偶联产物3,进而碱性条件脱除苯甲酰基,氢化脱除苄基,总共3步即可获得KRN7000,总收率高达57.3%。KRN7000的合成路线见图1。
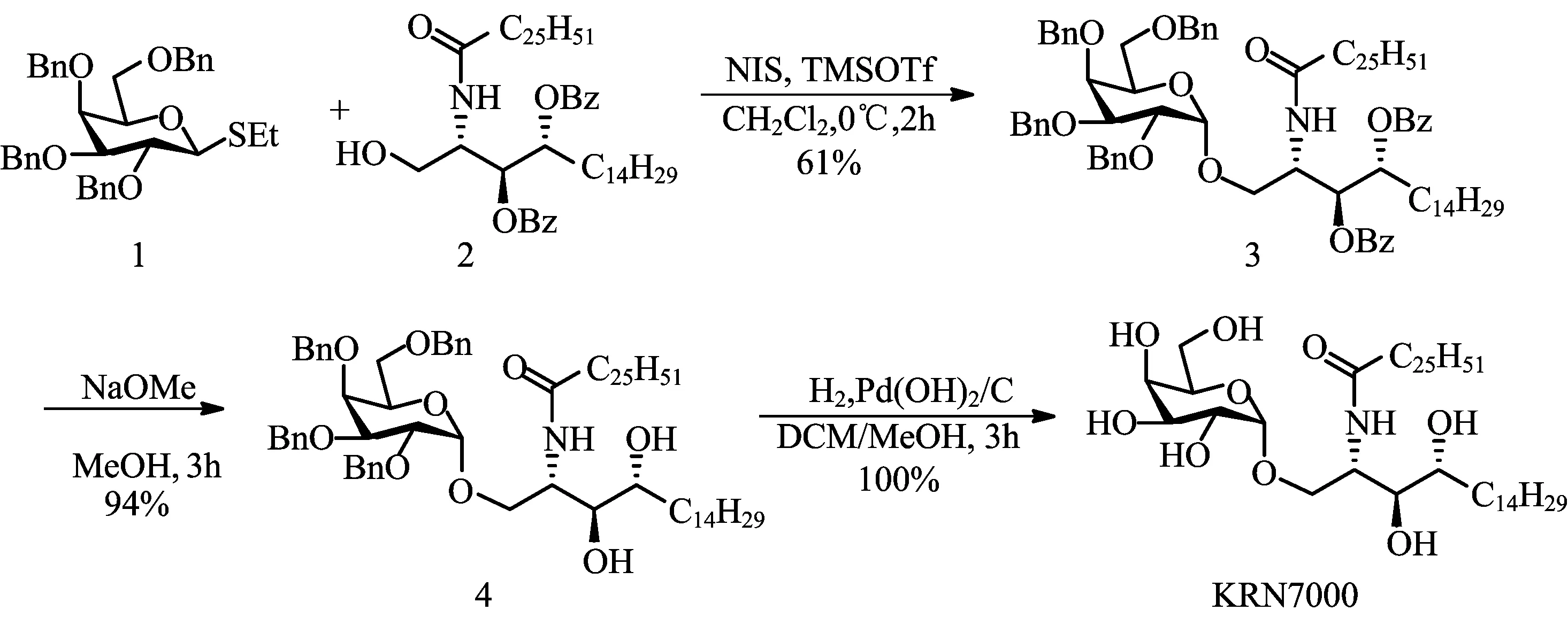
图1 KRN7000的合成路线
1.2.2化合物3的制备
将糖基供体1(1.31 g,2.25 mmol,1.5 eq)、受体2(1.35 g,1.5 mmol)和N-碘代丁二酰亚胺 (675 mg,4.5 mmol,2 eq)溶于干燥的二氯甲烷(5 mL)中,加入0.4 nm分子筛。在0 ℃下搅拌10 min,快速加入三氟甲基磺酸三甲基硅酯(TMSOTf,27 μL,0.15 mmol,0.1 eq),反应液持续在0 ℃搅拌2 h。加入三乙胺中和反应体系,加入饱和Na2S2O3溶液,反应用二氯甲烷萃取3次,减压浓缩,所得浓缩物用柱色谱分离,得到化合物3(构型为α),为无色液体(1.31 g,61%)。糖基化总收率为81%(α/β=3/1)。1H NMR(400 MHz,CDCl3):δ=8.03(d,J=7.2 Hz,2H),7.92(d,J=7.2 Hz,2H),7.59(t,J=7.2 Hz,1H),7.51(t,J=7.2 Hz,1H),7.45(t,J=7.2 Hz,2H),7.38~7.29(m,10H),7.27~7.17(m,12H),7.03(d,J=9.2 Hz,1H),5.70(dd,J=2.0,9.6 Hz,1H),5.42~5.38(m,1H),4.87(d,J=11.6 Hz,1H),4.75~4.72(m,2H),4.67~4.65(m,3H),4.56~4.50(m,3H),4.41(d,J=11.6 Hz,1H),4.07(t,J=5.6 Hz,1H),3.98(dd,J=2.0,10.4 Hz,1H),3.94(dd,J=2.4,12.0 Hz,1H),3.87~3.82(m,2H),3.61(d,J=10.0 Hz,1H),3.50(t,J=10.4 Hz,1H),3.22(dd,J=5.6,9.2 Hz,1H),2.16(t,J=7.6 Hz,2H),1.91~1.87(m,2H),1.64~1.60(m,2H),1.26~1.20(m,68H),0.88(t,J=6.0 Hz,6H)ppm;13C NMR(100 MHz,CDCl3)δ=173.1,166.1,165.3,138.8,138.4,138.3,137.8,133.2,132.8,130.2,130.0,129.8,128.5,128.3,128.2,128.1,128.0,127.9,127.7,127.6,127.5,127.4,127.3,100.1,78.7,75.0,74.7,73.9,73.5,73.2,72.8,72.3,70.3,70.0,69.1,48.6,36.7,31.9,29.7,29.6,29.5,29.4,29.3,28.4,25.7,22.7,14.1 ppm。
1.2.3化合物4的制备
将化合物3(830 mg,0.58 mmol) 溶于5 mL无水甲醇中,加入1 mol/L甲醇钠的甲醇溶液,调节pH值到9~10,然后在室温下搅拌3 h直到反应结束。加入酸性树脂IR-120中和,并加入过量的二氯甲烷使产物完全溶解,过滤,滤液减压蒸馏除去溶剂,所得浓缩物用快速柱色谱分离,得到化合物4为无色液体(660 mg,94%)。1H NMR(400 MHz,CD3OD/CDCl3=5/1)∶δ=7.39~7.26(m,20H),6.40(d,J=8.4 Hz,1H),4.94~4.85(m,3H),4.80~4.73 (m,2H),4.68(d,J=11.6 Hz,1H),4.57(d,J=11.6 Hz,1H),4.48(d,J=11.6 Hz,1H),4.39(d,J=11.6 Hz,1H),4.22(dd,J=3.6,8.0 Hz,1H),4.05(dd,J=3.6,9.6 Hz,1H),3.97(s,1H),3.89~3.85(m,4H),3.80(d,J=8.0 Hz,1H),3.53~3.48(m,4H),2.36(d,J=4.4 Hz,1H),2.13(t,J=7.6 Hz,2H),1.60~1.57(m,3H),1.49~1.42(m,1H),1.31~1.26(m,68H),0.89(t,J=6.8 Hz,6H)ppm;13C NMR(100 MHz,CD3OD/CDCl3=5/1)δ=173.2,138.5,138.4,137.9,137.6,128.5,128.4,128.3,128.2,128.1,128.0,127.9,127.7,127.6,127.5,99.2,79.3,76.2,76.1,74.8,74.5,74.2,73.6,73.3,72.8,70.0,69.9,69.0,49.6,36.8,33.3,31.9,29.7,29.6,29.4,29.3,25.9,25.7,22.7,14.1 ppm。
1.2.4KRN7000的制备
2 结果与讨论
2.1 糖基化反应
此路线的关键步骤是糖基供体1和受体2之间的糖基化反应,我们对此步进行优化,首先在25 ℃进行反应的尝试,实验结果显示只有痕量的产物3生成。因此降温至0 ℃,收率能达到21%。由于半乳糖部分原料相对于植物鞘氨醇部分价格便宜得多,因此定化合物2为1个当量,通过增加化合物1、NIS及TMSOTf的当量,发现收率有所提高(入口2~5)。最后确定最佳糖基化条件为n(2)∶n(1)∶n(NIS)∶n(TMSOTf)=1∶1.5∶2∶0.1,温度0 ℃。
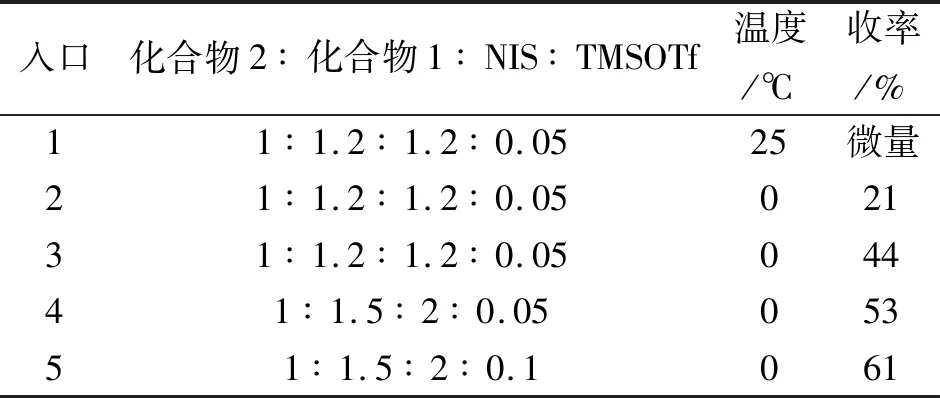
表1 投料比及温度对糖基化反应的影响
2.2 脱保护反应
最后一步脱除苄基的反应,我们尝试了几种溶剂,最终确定在二氯甲烷和甲醇的混合溶剂中能定量反应,只需要过滤出氢氧化钯/碳,蒸干即得高纯度的目标化合物KNR7000。
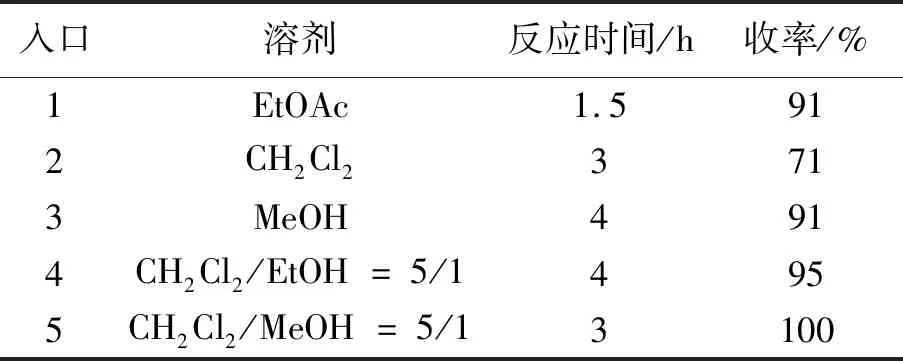
表2 溶剂对脱苄基反应的影响
3 结论
本文开发了一条以2,3,4,6-四-氧-苄基-α-D-半乳吡喃糖硫苷和苯甲酰基保护的植物鞘氨醇为起始原料,依次通过酸催化的糖苷化反应、碱性条件脱除苯甲酰基和氢氧化钯/碳通入氢气脱除苄基得到药物分子KRN7000。其中硫苷作为稳定易得的糖苷供体,使得糖苷化反应变得温和、易于操作。此路线总共3步,总收率达57.3%。该路线具有原料易得、操作简单、收率高等优点,为KRN7000类似物的合成提供了一种新的方法。
猜你喜欢
无机化学学报(2022年9期)2022-09-16
农业工程学报(2022年5期)2022-06-22
化工时刊(2022年1期)2022-05-25
作物学报(2022年7期)2022-05-12
复旦学报(医学版)(2021年5期)2021-10-13
药学进展(2021年3期)2021-05-11
药学进展(2021年3期)2021-05-11
井冈山大学学报(自然科学版)(2021年1期)2021-03-05
癌症进展(2018年11期)2018-12-30
药学研究(2015年12期)2015-12-08
