
β-环糊精6位修饰衍生物的研究进展
2021-03-29 02:07李紫沁张玉玲
化工技术与开发 2021年3期
李紫沁,贺 颖,姚 辉,韩 雪,张玉玲,邱 能
(成都理工大学材料与化学化工学院化工制药系,四川 成都 610059)
环糊精(Cyclodextrin)简称CD ,是由环糊精葡萄糖转移酶作用于淀粉、糖原、寡聚糖等葡萄糖聚合物而形成的一组环状低聚糖[1]。CD能与多种化合物形成包合物,从而提高包合物中客体分子的水溶性、稳定性和抗氧化、抗光解能力,降低毒副作用,同时能使客体分子达到缓释的效果[2]。
环糊精主要有α、β 、γ 三类[3-4],具有独特的空腔结构,并且内部疏水。脂溶性药物进入空腔后,依赖范德华力或氢键与CD形成稳定包合物后[5],能够调节药物的释放速度,提高药物的生物利用度[6]。其中β-CD空腔的内径大小适中,应用最为广泛[7],但其水溶性较低,在强酸环境下易裂解[8],因缺乏与金属离子配位的键合位点,不能对金属离子进行识别,从而限制了β-CD的应用。
要改善β-CD的性能,拓展其应用范围,可在2-、3-、6-羟基上进行可变取代。由于6-羟基的反应活性最高,目前研究最多的是6位羟基取代[9]。与β-CD相比,6位单取代衍生物可以增强药物的包合作用,更适合作为药物载体、稀释剂等[10]。本文就近年来β-CD的6位酯类衍生物、醚类衍生物、氨基类衍生物、酰胺类衍生物等的制备方法进行了总结。
1 β-CD的6位单取代
1.1 β-CD 的6位单取代酯类衍生物
β-CD的 6位单取代酯类衍生物是指β-CD的第6位羟基发生酯化反应后形成的酯类衍生物。β-CD的 6位单取代酯类衍生物[11-12]具有取代易定位、结构可控、易于分离等优势。
1.1.1 单-6(3-丙烯醛)-β-CD
许志刚等[13]以β-CD为母体化合物,在碱性水溶液中与丙烯酰氯反应,得到单-6(3-丙烯醛)-β-CD,该反应的产率为62.5%。该合成具有制备简单、反应易控制的优点,且产物有着良好的水溶性。合成路线见图1。

图1 单-6(3-丙烯醛)-β-CD的合成路线
1.1.2 单-6-脱氧-对甲苯磺酰基-β-CD
孙静等[14]将乳状的β-CD与对甲苯磺酰氯在碱性环境下反应3h,用盐酸调节pH至7,过滤除去不溶物后滤液冷藏。过滤后,用蒸馏水溶解后重结晶,真空干燥得到产物,产率为33.2%。合成方法虽然产率较低,但操作简单安全,且能够回收未反应的对甲苯磺酰氯。韩清华等[15-16]将β-CD溶解于碱性溶液中,先加入对甲苯磺酰氯反应2h,再加入对甲苯磺酰氯反应3h。之后抽滤分离产物,产率达到46.2%。

图2 单-6-氧-对甲苯磺酰基-β-CD的合成路线
1.1.3 乙氧羰基化-β-CD
张毅民等[17]在碳酸钾的作用下,用碳酸二乙酯与β-CD发生反应,合成乙氧羧基化-β-CD。由实验可知,在DEC与β-CD的质量比20.2∶1、反应8.5h的条件下,产率最高为98%。合成路线见图3。

图3 乙氧羰基化-β-CD的合成路线
相较于传统的合成反应采用有毒的羧基化试剂[18],该合成反应使用的试剂DEC相对更加绿色安全。该合成法利用工艺模型,研究了不同因素对收率的影响,从而优化了合成路线。
1.1.4 十二烯基琥珀酸CD酯
安英杰等[19]将β-CD溶于蒸馏水后加入碳酸钠,并调节pH至8.5,加入十二烯基琥珀酸酐(DDSA)充分反应后,调节pH至中性,用乙醇水溶液萃取、洗涤、抽滤、干燥后,得到十二烯基琥珀酸CD酯,合成路线见图4。该产品在食品、日化、微胶囊等领域具有广泛的应用前景。

图4 十二烯基琥珀酸CD酯的合成路线
1.1.5 顺丁烯二酸-β-CD酯
李咏富等[20]采用半干反应法,将β-CD、顺丁烯二酸、次亚磷酸钠依次加入耐压瓶内,加入阻聚剂4-甲氧基酚充分反应后,得到顺丁烯二酸-β-CD酯。合成路线见图5。

图5 顺丁烯二酸-β-CD酯的合成路线
1.2 β-CD的6位单取代醚类衍生物
β-CD的6位单取代醚类衍生物是指β-CD第6位羟基与另一个含有羟基的化学物质发生偶联反应,变为醚键。β-CD的醚类衍生物的溶解性显著增强。目前该类衍生物广泛应用在药物制剂和分离科学等领域。
1.2.1 磺丙基醚-β-CD
李夏阳等[21]为了解决传统合成方法中的操作危险、副产物多、产物不纯等问题,设计了3种新型合成路线(图6)。
三步合成法:以β-CD和1,3-苯磺内酯为原料,在水浴加热条件下,分3次加入碱,并严格控制碱的用量,得到磺丙基醚-β-CD。
微波协同合成法:微波辐射手段可使能量分布均匀,避免了高温对产物的分解,并且显著缩短反应时间。此方法可以有效减少副产物生成,从而提高产物收率。
碱性电解水催化合成法:将β-CD完全溶解于碱性电解水相中,在较低功率下反应后,再调回原功率反应,得到均相产品液。

图6 磺丙基醚-β-CD的合成路线
1.2.2 磺丁基醚-β-CD
张毅民等[22]将β-CD溶于碱性溶液中与1,4-丁磺内酯反应,将pH控制在9~11,反应完成后调节pH至中性。反应液经脱盐、浓缩及分离纯化后,经喷雾干燥得到磺丁基醚-β-CD,产率达到85%,合成路线见图7。该反应是在水相体系中采用分步加碱的方式进行,过程用碱量少,条件温和,后处理简单,符合绿色工艺发展的特点。

图7 磺丁基醚-β-CD的合成路线
1.2.3 磺烷基醚-β-CD
宋爱晶等[23]将氢氧化钠溶于水中制备成高碱溶液,再加入β-CD,剧烈搅拌后与磺内酯反应。用盐酸将pH调至中性,滤液经冷冻干燥后得到磺烷基醚-β-CD。合成路线见图8。

图8 磺烷基醚-β-CD的合成路线
1.2.4 胺乙基-β-CD
刘慧君[24]采用2-氯乙胺盐酸盐在碱性环境中与β-CD反应的方法,合成了胺乙基-β-CD[25]。合成路线见图9。该方法安全,简单易行,所用时间少,成本低,提高了利用价值。

图9 胺乙基-β-CD的合成路线
1.3 β-CD 的6位单取代氨基类衍生物
β-CD的 6位单取代氨基类衍生物是指-CD的第6位羟基发生了基团转化,变成氨基,形成了氨基类衍生物。经过氨基修饰的β-CD活性增强,有利于后续进一步的修饰,β-CD氨基类衍生物可以作为一个中间体,得到最后需要的产品。
1.3.1 单-6-脱氧-6-氨基-β-CD
Vijay Vilas Shinde等[26]将β-CD与甲苯磺酰咪唑在碱性条件下反应后,再加入氯化铵,得到单-6-脱氧-邻甲苯磺酰基-β-CD,然后加入氨水继续反应,得到单-6-脱氧-6-氨基-β-CD,总收率为39.49%,合成路线见图10。该反应步骤简单,反应物安全,但是产率较低。修饰后的β-CD价格便宜,稳定性高,可重复使用,可以作为超分子有机催化剂。

图10 单-6-脱氧-6-氨基-β-CD的合成路线
Temimi等[27]将单-6-氧-对甲苯磺酰基-β-CD溶解在80℃水中回流过夜,冷却至室温后,倒入丙酮中离心,真空干燥一夜,得到产物单-6-氧-对甲苯磺酰基-β-CD,再加入叠氮化钠、甲酸铵反应,得到单-6-脱氧-6-氨基-β-CD,合成路线见图11。

图11 单-6-脱氧-氨基-β-CD的合成路线
1.3.2 叶酸-胺基-β-CD
韩彬等[28]将叶酸溶解于N,N-二甲基甲酰胺中,加入NHS和DCC搅拌,随即向反应液中加入单-6-氨基-β-CD和吡啶,充分反应后得到产物叶酸-胺基-β-CD。合成路线见图12。
1.4 β-CD的 6位羟基被OTS活化后的单取代衍生物
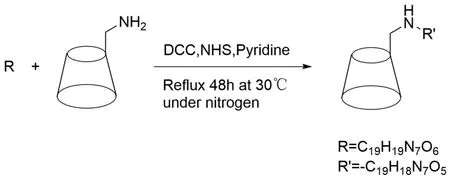
图12 叶酸-胺基-β-CD的合成路线
β-CD 的6位羟基活性较大,易发生取代,用OTS活化β-CD,可得到单-6-脱氧-对甲苯磺酰基-β-CD,然后再与含氨基的化合物进行反应,可得到氨基类衍生物,其水溶性增强,应用广泛。
1.4.1 单(6-乙二胺-去氧)-β-CD
冯锋等[29]将单-6-氧-对甲苯磺酰基-β-CD[30-31]加入乙二胺中,搅拌溶解后升温至80℃,反应后减压蒸除溶剂。将所得固体溶解在热水中,搅拌的同时加入丙酮,过滤收集白色沉淀,提纯,得到单(6-乙二胺-去氧)-β-CD,产率为80.3%,合成路线见图13。

图13 单(6-乙二胺-去氧)-β-CD的合成路线
张淼等[32]使用相同的原料进行反应,唯一不同的是在75~80℃温度下反应,该反应的产率为81.5%。
1.4.2 叶酸-乙二胺基-β-CD
韩彬等[28]将叶酸溶解于N,N-二甲基甲酰胺中,再加入NHS和DCC并搅拌,随即向反应液加入单-6-脱氧-乙二胺-β-CD和吡啶发生反应,生成叶酸-乙二胺基-β-CD。

图14 叶酸-乙二胺基-β-CD的合成路线
1.4.3 UPA -β-CD
赵楠等[33]以DMF为溶剂,6-乙二胺-β-CD和马来酸酐为原料,反应4h后,用三氯甲烷沉淀,丙酮洗涤,干燥后得到UPA-β-CD,产率为70%,合成路线见图15。该产物在酸性环境下的溶解性强,手性拆分方便。
1.4.4 单6-丙烯酰化丁香酸-β-CD
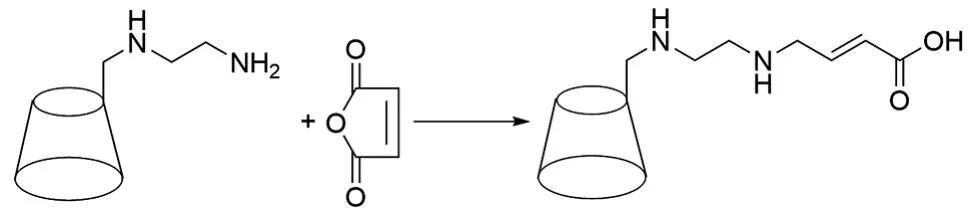
图15 UPA-β-CD的合成路线
朱玭玭等[34]将单-6-乙二胺-β-CD、丙烯酰化丁香酸和1-羟基苯并三唑溶解于DMF中,加入N,N-二异丙基乙胺 DIPEA和 1- (3-二甲氨基丙基) -3-乙基碳二亚胺盐酸盐,反应后经抽滤、纯化、干燥,得到单6-丙烯酰化丁香酸-β-CD,产率为80%,合成路线见图16。该方法具有反应彻底、产物易分离提纯、产率高的优点。

图16 单6-丙烯酰化丁香酸-β-CD的合成路线
1.4.5 生物素修饰的β-CD
Yong Chen等[35]将生物素和1-羟基苯并三唑溶解于干燥的DMF中,在冰浴中搅拌0.5h,然后加入单-6-脱氧-6-乙二胺-β-CD和N,N’-二环己基碳二亚胺,再缓慢滴加干燥的DMF,干燥后得到生物素修饰的β-CD,合成路线见图17。

图17 生物素修饰的β-CD合成路线
1.4.6 单6-丙烯酰乙二胺-β-CD
朱玭玭等将单6-乙二胺β-CD溶解在1-羟基苯并三唑中,再依次加入丙烯酸、N, N-二异丙基乙胺 和 1- (3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,反应后经抽滤、纯化、干燥,得到单6-丙烯酰乙二胺-β-CD,产率为88%,合成路线见图18。与传统路线[36]相比,本反应的效率得到了显著提升。

图18 单6-丙烯酰乙二胺-β-CD的合成路线
1.4.7 单-6-脱氧-二乙烯三胺-β-CD
韩彬等[28]将单-6-氧-对甲苯磺酰基-β-CD在75℃下与干燥处理后的乙二烯三胺反应,生成单-6-脱氧-二乙烯三胺-β-CD,产率为70%,合成路线见图19。
1.4.8 叶酸-二乙烯三胺基-β-CD

图19 单-6-脱氧-二乙烯三胺-β-CD的合成路线
张淼等[32]在氮气保护下,将叶酸在二甲基亚砜中充分溶解后,加入NHS和DCC,在30℃避光搅拌2h,再加入用干燥的吡啶溶解的单-6-脱氧-二乙烯三胺-β-CD,继续在30℃下避光反应40h。结束后过滤不溶副产物DCU,减压蒸馏去除吡啶。丙酮中析出产物,过滤得到粉末状固体,用丙酮洗涤,45℃真空干燥,得到叶酸-二乙烯三胺基-β-CD,合成路线见图20。

图20 叶酸-二乙烯三胺基-β-CD的合成路线
1.4.9 单-6-脱氧-三乙烯四胺-β-CD
韩彬等将单-6-氧-对甲苯磺酰基-β-CD在75℃下与干燥处理后的三乙烯四胺反应,生成单-6-脱氧-三乙烯四胺-β-CD,产率为70%,合成路线见图21。

图21 单-6-脱氧-三乙烯四胺-β-CD的合成路线
1.4.10 叶酸-三乙烯四胺基-β-CD
韩彬等[28]将叶酸溶解于N,N-二甲基甲酰胺中,再加入NHS和DCC搅拌,随即向反应液中加入单-6-脱氧-三乙烯四胺-β-CD和吡啶,充分反应后得到叶酸-三乙烯四胺基-β-CD,合成路线见图22。

图22 叶酸-三乙烯四胺基-β-CD的合成路线
1.5 β-CD的 6位单取代氨基酸类衍生物
β-CD的6位羟基与氨基酸反应,得到含氨基的化合物修饰的衍生物,再加入化合物对氨基进行修饰,得到β-CD的6位单取代氨基修饰类衍生物,增强了其在酸性溶剂中的水溶性。
1.5.1 L-精氨酸-β-CD
赵美霞等[37]将L-精氨酸、单-(6-O-对甲苯磺酰基)-β-CD、水和三乙醇胺的混合溶液在氮气保护下,加热到85℃反应24h,冷却重结晶后得到产物,产率为35.2%,合成路线见图23。此方法将氨基酸与CD进行连接,能用于研究水溶液中的分子识别、包合作用实质、分子的自组装等内容[38]。

图23 L-精氨酸-β-CD的合成路线
1.5.2 单-(6-L-氨基酸-6-脱氧)-β-CD
吴磺溢等[39-40]以单-(6-O-对甲苯磺酰基)-β-CD为原料,与L-丝氨酸、L-苏氨酸和L-酪氨酸发生亲核取代反应,得到了3种氨基酸改性的β-CD衍生物单-(6-L-氨基酸-6-脱氧)-β-CD,其产率分别为22%、25%、29%,合成路线见图24。

图24 单-(6-L-氨基酸-6-脱氧)-β-CD的合成路线
1.6 β-CD的6位单取代酰胺类衍生物
1.6.1 α-FACD、γ-FACD、FA-diCD
Juan-Juan Yin等[41]在叶酸的混合物中加入DCC、NHS、DMF,在黑暗中保持3h后加入单-6-脱氧-6-氨基-β-CD、吡啶进行反应。反应后过滤得到沉淀物,用乙腈、乙酸乙酯和乙醇洗涤后,得到3种产品α-FACD、γ-FACD、FA-diCD,产率分别为41.6%、11.4%、7.1%,合成路线见图25。

图25 α-FACD、γ-FACD、FA-diCD的合成路线
1.6.2 双氯芬酸-β-CD
Vieira等[42]采用两步法合成,首先对β-CD甲酰化,得到单-6-脱氧-对甲苯磺酰基-β-CD,然后在微波下对双氯芬酸钠进行核物质取代,得到双氯芬酸-β-CD。合成路线见图26。

图26 双氯芬酸-β-CD的合成路线
1.7 其它类β-CD的6位单取代衍生物
1.7.1 单-6-去氧-碘代-β-CD
张淼等将单-6-脱氧-对甲苯磺酰基-β-CD和碘化钾放入圆底烧瓶,加入DMF加热搅拌,在90℃反应5h后,减压蒸馏除去溶质,用丙酮除去未反应完的KI,抽滤得到固体,80℃下真空干燥12h,得到单-6-去氧-碘代-β-CD,产率为81%,合成路线见图27。

图27 单-6-去氧-碘代-β-CD的合成路线
1.7.2 6-氧-烯丙基-β-CD
朱玭玭等将β-CD溶于无水DMF中,冰水浴下加入氢化钠后搅拌1h,再逐滴加入3-溴丙烯,室温反应7h。将反应液分散在丙酮中,抽滤、干燥后得到6-氧-烯丙基-β-CD,合成路线见图28。利用亲核反应来合成以双取代为主的6-氧-烯丙基-β-CD,方法操作简单,耗时短。

图28 6-氧-烯丙基-β-CD的合成路线
1.7.3 手性吡咯烷官能团修饰β-CD
翟琳等[43]在氮气保护下,将单-(6-脱氧-对甲苯磺酰基)-β-CD悬浮于干燥的DMF中,升温后加入手性吡咯烷,侧链反应后加入氢氧化钠,再将混合物倒入丙酮溶液中,析出白色沉淀,过滤后得到手性吡咯烷官能团修饰的β-CD[44],产率为67%,合成路线见图29。
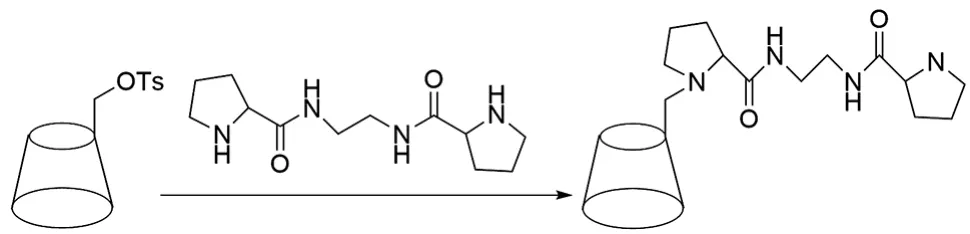
图29 手性吡咯烷官能团修饰β-CD的合成路线
1.7.4 单-(6-邻氨基苯甲酸-6-脱氧)-β-CD
周兴龙等[45]在氮气保护下,将DMF、邻氨基苯甲酸、碳酸钠和单-(6-脱氧-对甲苯磺酰基)-β-CD在85℃下反应12h后,用丙酮洗涤、DMF洗脱后得到产物,产率为82%,合成路线见图30。产物能够在水相中高效催化Heck偶联反应,能应用于绿色环保催化反应中。

图30 单-(6-邻氨基苯甲酸-6-脱氧)-β-CD的合成路线
2 结论
利用化学方法对β-CD 的6位进行修饰,可得到一系列的β-CD衍生物,从而改善CD的理化性质,扩大其在食品、环境、化工、农药、医药学等领域的应用范围[46]。目前对CD的修饰方法有[47-48]:①直接修饰羟基;②将羟基选择性地转变为高亲核活性的基团,再进行修饰;③将羟基选择性地转变为可离去基团,再进行修饰;④其他特殊化修饰。
对β-CD 的6位羟基进行修饰,可提高β-CD的水溶性和包合能力,扩大β-CD及其衍生物在生物医药方面的应用[49]。β-CD及其衍生物的包合药物可用于治疗阿尔兹海默症、癌症等多种疾病。
目前6-β-CD的衍生物种类繁多,主要包含醚类、酯类、氨基类和酰胺类等。研究者开发了各种制备方法,但是这些制备方法很多还不够成熟,存在产率较低、制备周期长、所用试剂危险性较大等问题。因此对β-CD进行修饰,需要进一步对其制备方法进行改进,以提高产率,找到更绿色、环保、安全的制备方法。随着社会的发展,人们对高端医药产品的需求越来越高,因此β-CD衍生物的应用将会越来越广泛。
猜你喜欢
皮革制作与环保科技(2022年14期)2022-08-28
农业工程学报(2021年3期)2021-04-15
陶瓷学报(2020年6期)2021-01-26
中华养生保健(2020年9期)2021-01-18
石油化工技术与经济(2020年4期)2020-09-15
茶叶(2020年1期)2020-05-30
山东农业科学(2019年9期)2019-12-09
世界农药(2019年3期)2019-09-10
无机化学学报(2019年2期)2019-02-27
中学生数理化·高二版(2016年3期)2016-12-26
