
基于RAD高通量测序的贵州百里杜鹃保护区杜鹃花属分类*
2021-04-02 04:40黄承玲田晓玲任永权黄家湧马永鹏
林业科学 2021年2期
黄承玲 姚 刚 田晓玲 任永权 黄家湧 马永鹏
(1.贵州民族大学生态环境工程学院 贵阳 550025; 2.云南省极小种群野生植物综合保护重点实验室 中国科学院昆明植物研究所昆明 650201; 3.贵州民族大学人文科技学院 贵阳 550025; 4.贵州百里杜鹃科研所 毕节 551614)
贵州省百里杜鹃保护区位于贵州西北部,其自然分布的杜鹃花属(Rhododendron)植物资源十分丰富,是国内为数不多的以高山杜鹃为观赏和保护对象的省级自然保护区、省级风景名胜区和国家级森林公园。近年来,百里杜鹃不仅成为热门的观花旅游目的地,还因其独特的杜鹃花资源成为科学研究的热点。杨成华等(2006)、Chen等(2010a)、张长芹等(2015)等都对百里杜鹃区域的杜鹃花资源进行过详细的调查研究,但具体到个别物种,尤其是近年发表的该区域的新种的分类地位问题仍存在争议(Marczewskietal.,2016)。以上研究对百里杜鹃保护区的杜鹃花属仅是依据形态学特性进行分类。比如,花芽的着生位置和叶背面是否有鳞片是区分亚属的重要形态特征; 而植株、花冠形状与颜色,以及叶片、花瓣、雌雄蕊毛的类型(比如:刚毛、绒毛、腺体毛、星状毛、杯状毛)是区分不同亚组和物种的重要特征(胡琳贞等,1991)。然而,形态学的一个突出问题是很难把握种间和种内形态变异的幅度(Marczewskietal.,2016)。因而,随着分子生物学技术的发展,有必要借助分子标记技术对百里杜鹃保护区杜鹃花属的分类及存疑进行印证和解答。目前对百里杜鹃的杜鹃花属系统分类的分子水平研究较少。何云松等(2015)采用叶绿体基因组的trnL-F间隔区测序,研究了百里杜鹃保护区26种杜鹃花属植物的系统发育关系,其结果显示trnL-F可以在亚属水平区分物种,而在物种水平仅有少数种被明确区分,说明杜鹃花属分类的复杂与困难。近年来第2代测序技术(高通量测序技术)因其单次运行产出的序列数据量大、价格低廉而广泛用于植物的遗传转化、分子育种研究中(时小东等,2016; 胡玉玲等,2014; 邓敏捷等,2013)。在第2代测序技术基础上,一项基于全基因组酶切位点的简化基因组测序技术RAD-seq(restriction-site associated DNA-sequencing)迅速发展起来,该技术通过对限制性酶切位点的标记进行测序,在大多数生物中获得海量的单核苷酸多态性(single nucleotide polymorphism,SNP)位点(Milleretal.,2007; Tasselletal.,2008; 桂柳柳,2017),因具有多态性位点数量多、分布密度高且操作方便等特点逐渐被应用到植物群体遗传学研究中(魏明科等,2018; 王洋坤等,2014)。李云飞等(2019)采用RAD-seq技术开发了85种杜鹃花属植物的高质量SNP位点,通过高通量测序探讨分类,在亚属水平的分类取得很好的效果。
百里杜鹃区域杜鹃花属中很多种亲缘关系近,自然杂交相当普遍(Zhangetal.,2007; Marczewskietal.,2016)。本研究首先剔除前期研究中证实为天然杂交的种(张长芹等,2015),采集该区域33种杜鹃花属植物进行高通量测序并进行系统分类,对个别物种的形态与系统分类结果不一致的原因进行探讨,以期实现百里杜鹃保护区杜鹃花属的准确分类,并为该区域杜鹃花资源的新品种选育提供指导。
1 材料与方法
1.1 试验材料
用于RAD-seq技术测序的杜鹃花属植物样本共34份,采样信息见表1,隶属于6个亚属7个组10个亚组(表2),其中大果杜鹃(Rhododendronglanduliferum)采自贵州盘县,其余33种按张长芹等(2015)的描述和记录,采自贵州省百里杜鹃自然保护区。每个种采集5株成年个体的健康幼嫩叶片2~3片,采样的个体间距10 m以上,采集的叶片保存于4 ℃冰箱备用。

表1 杜鹃花属34个种的采样信息Tab.1 Sample information of 34 species in Rhododendron
1.2 试验流程
每个个体摘取新鲜叶片0.5 g,用改良的十六烷基三甲基溴化铵(hexadecyl trimethyl ammonium bromide,CTAB)法分别提取样品的基因组DNA并进行DNA质量检测(Doyle,1987),对质量合格的DNA,参考Patterson 等(2012)流程进行建库; 用Illumina HiSeq 4000 System(San Diego,CA,USA)测序平台进行测序。
1.3 原始数据过滤
原始测序数据(raw data)中一般会有少部分的reads带有测序引物、接头等人工序列,为保证后续分析的准确性,需要先对raw data进行过滤,去除影响数据质量和后续分析的低质量区域(王成贺,2016)。本研究采用Stacks 2.5.2软件的process_radtags程序(Rochetteetal.,2017; 2019),按照barcode 序列和index序列对个体进行区分并作初步的质量控制,去除含有接头及低质量的序列,得到clean data,将所有得到的reads剪切为135 bp,然后通过FastQC 0.11.8 (Andrews, 2015) 软件进行个体reads的质量评估。
1.4 基于无参考基因组的变异检测
采用Stacks-2.5.2软件进行数据预处理,包括denovo组装和不连锁变异位点的过滤。Stacks分析流程如下:1)使用Linux系统的cat命令合并双端数据。2)采用ustacks根据序列间相似性原则和最大似然模型对每个样本进行denovo组装,将完全一致的序列聚为一个stacks,生成的结果为loci。具体使用的关键参数为:-M 5 -m 3 -R。3)使用cstacks对所有样本的loci进行合并,采用kmer的方法来进行比对,得到catalog.allels.tsv.gz,catalog.snps.tsv.gz和catalog.tags.tsv.gz文件和log文件。具体使用的关键参数为:-n 3。4)采用sstacks把所有样本的序列match到cstacks生成的catalog文件中,得到各个样本的*.matches.tsv.gz文件和log文件。此步骤采用默认参数。5)使用tsv2bam把sstacks生成的各样本的matches.tsv.gz文件转为各locus的bam文件,得到各个样本的*.matches.bam文件和log文件。此步骤采用默认参数。6)最后使用populations检测所有样本的SNPs,此步骤具体使用的关键参数为:--write-random-snp --vcf -plink,得到包含SNP的VCF格式文件。
1.5 系统发生树构建
首先采用VCFtools(Daneceketal.,2011)对上一步stacks得到的SNP进行过滤,所使用的关键参数包括:--min-alleles 2 --max-alleles 2 --remove-indels --maf 0.05 --max-missing 0.2,--minGQ 30 --minDP 3。得到42 083个SNP位点,对这些位点进行中性检验,所使用关键参数为--TajimaD 2500,参照Tajima’sD的95%置信区间表(Tajima,1989)筛选中性位点。在得到中性位点的VCF文件后,使用Tassel 5.0(Bradburyetal.,2007)将其转化为phy格式。对phy格式的SNP矩阵采用concatenation的最大似然法(Maximum Likelihood)进行系统发生分析,使用IQTREE(Minhetal.,2020; Kalyaanamoorthyetal.,2017; Hoangetal.,2018) 在北京超级云计算中心A分区上进行运算,从242个DNA碱基替换模型中筛选出的最佳碱基替换模型为TVM+F+ASC,以超快bootstrap法评估系统发生树分支的支持度,指定次数为1 000。对于IQTREE得到的系统发生树文件,使用FigTree(Priceetal.,2010)进行查看和美化处理。
1.6 群体遗传结构fastStructure分析
通过plink转换为二进制文件(得到.ped和.map文件),然后以转化好格式的SNP文件作为输入文件,设置1≤K≤10,对fastStructure(Aniletal.,2014)使用for-do语句进行群体聚类分析,然后使用grep提取最佳K值,即cross-validation error值最低时所对应的K值。设置个体Q≥0.9作为判断物种的标准。
1.7 主成分分析(PCA)
主成分分析是群体遗传学中常用的分析手段,目前用于主成分分析的数据主要为高密度的SNP标记,常使用的分析软件为GCTA(Jianetal.,2011),通过对获得的中性SNP位点的数据集进行矩阵转换,利用eigen函数计算所有样本遗传关系矩阵的特征值和特征向量,再使用R语言的ggplot2包进行分析结果的可视化,并根据检索得到的样品亚属信息进行分类。
2 结果与分析
2.1 测序数据特性
对杜鹃花属34个种的样本进行RAD-seq技术测序,共获得测序数据量15.1 Gb,平均每个个体455.58 Mb。使用fastp软件对原始数据进行质量控制,得到10.22 Gb的清洗数据,平均每个个体307.80 Mb。另外,数据的Q20、Q30也分别从过滤前的94.44%、87.11%提高到过滤后的97.43%、94.32%。数据产出、测序深度、覆盖度和多态性位点的具体信息见表2。
2.2 群体结构分析
混群分析结果显示,当K=4时,曲线出现最低值(交叉验证错误率cross-validation error=0.013 332 7); 同时,考虑到样本所归属的分类单元,K=3(cross-validation error=0.030 077 3)和K=5(cross-validation error=0.033 354 2)也做了群体结构柱状堆叠图(图1)。由图1可知:当K=3时,蓝色、绿色和红色代表3类不同的遗传组成,其中蓝色对应映山红亚属的全部,绿色对应常绿杜鹃亚属的大部分,红色则包含全部的杜鹃亚属、糙叶杜鹃亚属、马银花亚属和羊踯躅亚属,并包含部分常绿杜鹃亚属个体; 当K=4时,马银花亚属与羊踯躅亚属和有鳞类杜鹃(杜鹃亚属和糙叶杜鹃亚属)分开,而常绿杜鹃亚属所有种也与有鳞类杜鹃完全分开; 当K=5时,分辨率降低,马银花亚属、羊踯躅亚属和有鳞类杜鹃无法区分,而常绿杜鹃亚属内部分为3个部分,但与映山红亚属无法区分。综合来看,K=4时可将5个亚属中的4个亚属很好地区分,且支持形态证据对这些种的划分; 而常绿杜鹃亚属内部分为2组反映了该亚属复杂的系统进化关系,映山红亚属无法与部分常绿杜鹃亚属区分说明二者具有较近的亲缘关系。
2.3 SNP位点主成分分析
PCA 聚类结果(图2)与fastStructure结果(图1)相似,34个杜鹃花属植物样本聚成5类,即常绿杜鹃亚属、羊踯躅亚属、马银花亚属和映山红亚属的各自物种聚在一起,与基于形态性状的划分完全吻合; 糙叶杜鹃亚属与杜鹃亚属聚在一起,反映了2个亚属亲缘关系较近。
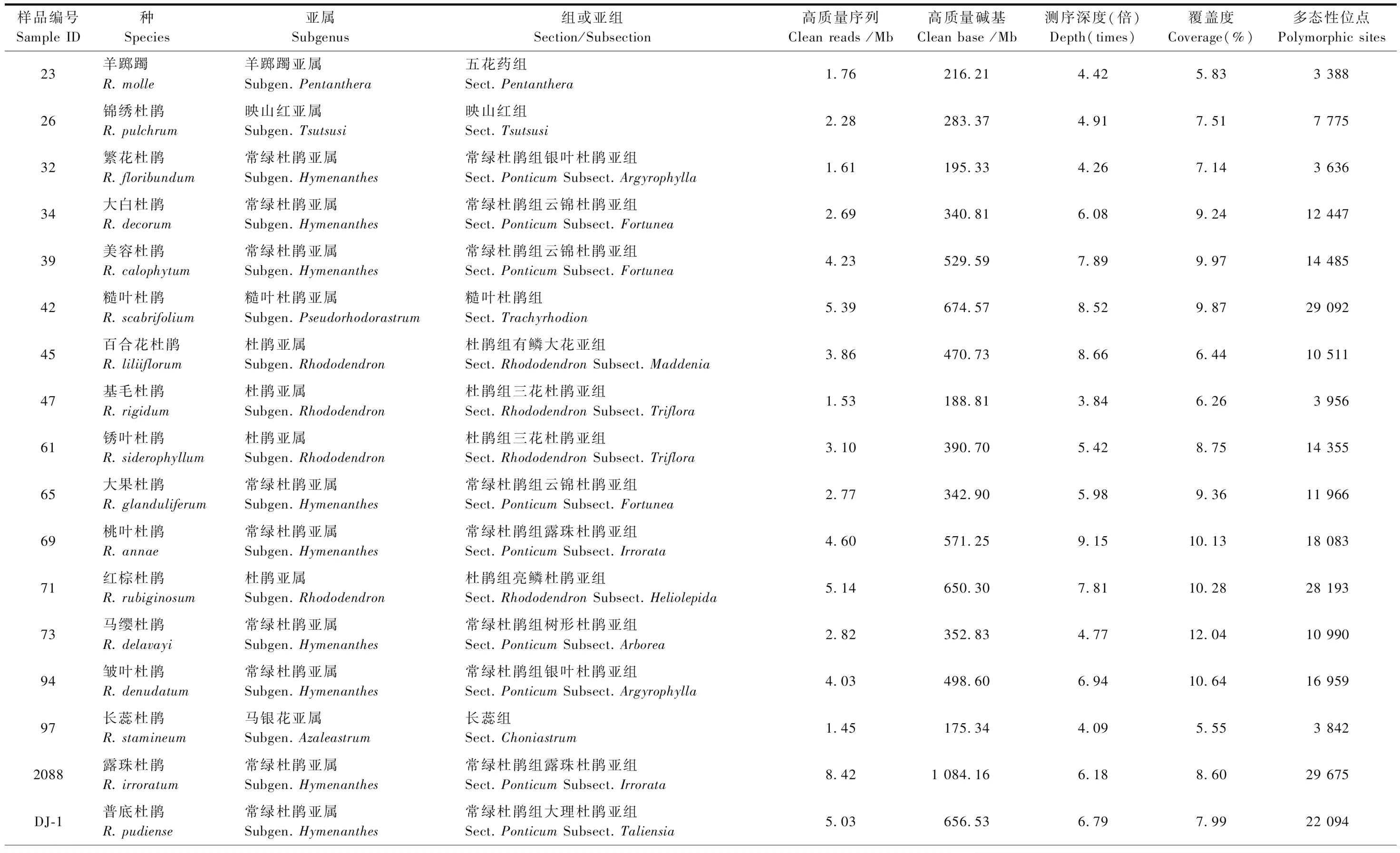
表2 杜鹃花属34个种RAD测序的数据特性Tab.2 Data characteristics of 34 species in Rhododendron by RAD sequencing

续表2 Continued
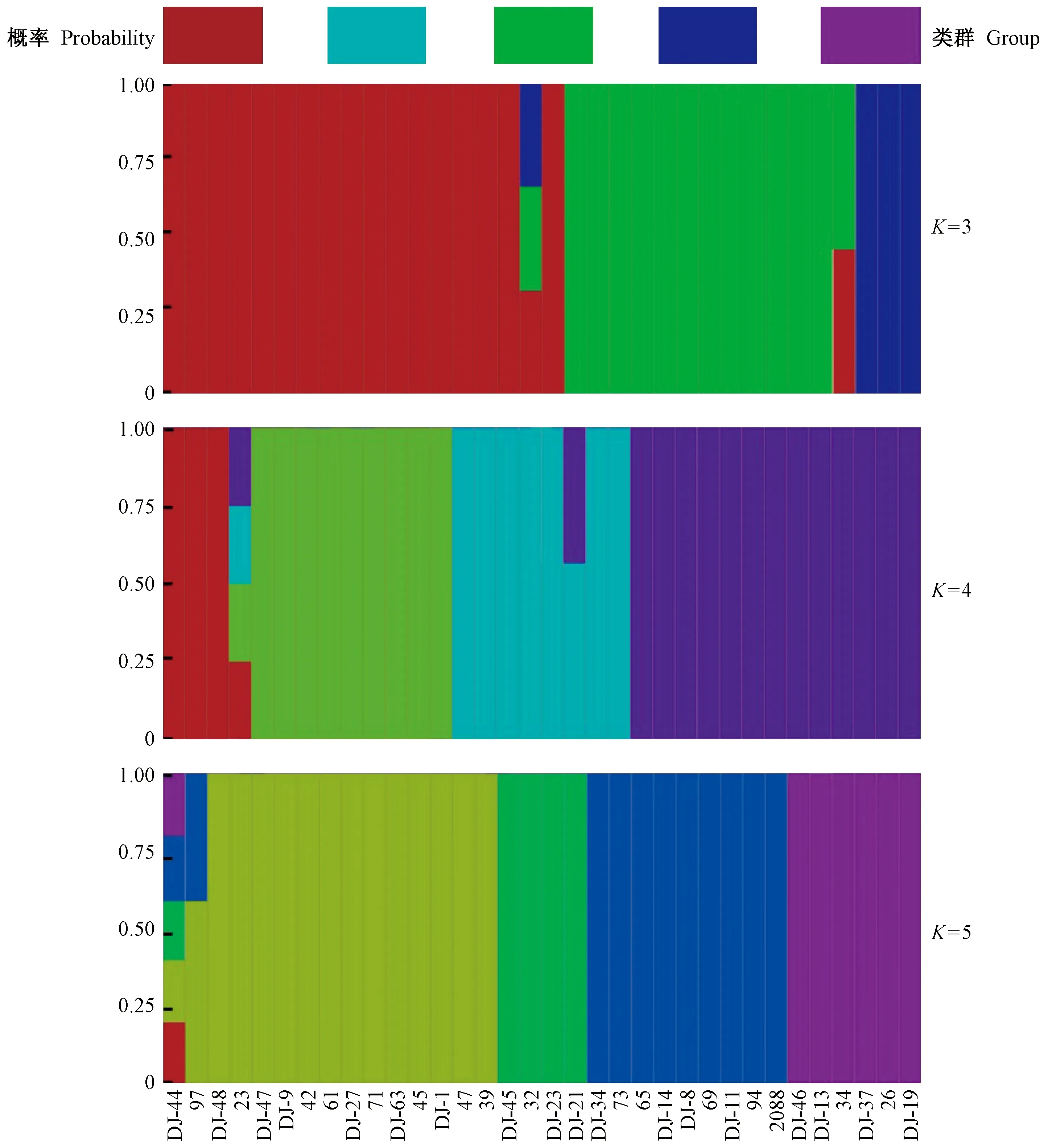
图1 基于28 983个SNP位点的杜鹃花属34个种的fastStructure聚类Fig. 1 Results from fastStructure analysis of 34 Rhododendron species based on 28 983 SNPs

图2 基于28 983个SNP位点的杜鹃花属34个种的PCA聚类Fig. 2 Results from PCA analysis of 34 Rhododendron species based on 28 983 SNPs
2.4 系统发生树构建
采用IQ-Tree软件对杜鹃花属34个种的28 983个单核苷酸多态性位点(SNPs)构建系统发生树,使用筛选好的模型TVM+F+ASC进行最大似然亲缘关系分析(maximum-likelihood tree),结果(图3)与PCA主成分分析(图2)相似,即34个种的样本聚成5支,其中4支分别对应形态上其各自归属的常绿杜鹃亚属、羊踯躅亚属、马银花亚属和映山红亚属,同样,糙叶杜鹃亚属与杜鹃亚属聚成1支,反映了2个亚属亲缘关系较近。一些形态极其相似的种无法很好地区分开来,比如九龙山杜鹃和大果杜鹃,前者大部分性状如幼枝被刚毛状腺体、叶形状与大小、叶柄有腺体等与《中国植物志》中大果杜鹃的形态描述都有重叠(胡琳贞等,1991); 而繁花杜鹃与皱叶杜鹃这2个种的差别仅在于叶背面毛被的颜色。
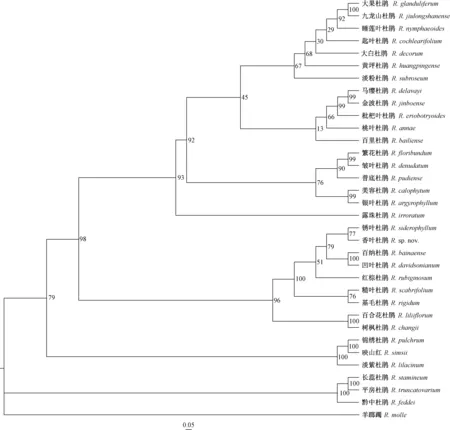
图3 基于28 983个SNP位点的杜鹃花属34个种的系统发生树Fig. 3 Results from phylogenomic analysis of 34 Rhododendron species based on 28 983 SNPs
3 讨论
通过对百里杜鹃保护区杜鹃花属34个种的简化基因组测序,以获得的高质量SNP位点为基础,从基因组水平系统分析了34个种的系统演化关系。从fastStructure、PCA聚类和系统发生树的结果来看,常绿杜鹃亚属可以与其他5个亚属完全分开独立为一支,支持率92%,这与Kurashige等(2001)、何云松等(2015)、李云飞等(2019)的结论一致,表明常绿杜鹃亚属是一个单系类群。而叶片无鳞的马银花亚属和映山红亚属聚成一个分支,表明二者亲缘关系较近; 叶片同样无鳞的羊踯躅亚属独立成一支。糙叶杜鹃亚属和杜鹃亚属不能完全分开,这与Kurashige等(2001)、李云飞等(2019)的结论一致,这2个亚属属于叶片有鳞类群,印证了李云飞等(2019)在亚属水平可以用叶片被鳞与否来解释基因组水平各亚属的系统分类学关系的结论。
从组、亚组水平看,常绿杜鹃亚属包含的各个亚组的多个种交错组成3个分支,这个结果与Kurashige 等(2001)、何云松等(2015)、李云飞等(2019)的研究一致,表明各亚组之间的界限模糊; 对该亚属不同亚组植物的天然杂交现象进行研究(Zhangetal.,2007; Zhaetal.,2010; Yanetal.,2013; Maetal.,2016; 2010; Marczewskietal.,2016),结果表明分布在同一个区域内的常绿杜鹃亚属不同亚组之间的自然杂交极为频繁,很多种类出现杂交渐渗、基因水平转移以及不完全谱系分化等现象,使得常绿杜鹃亚属的亚组以下物种水平的种间关系很难准确界定(Milneetal.,2010; Yanetal.,2015)。
从物种水平看,张长芹等(2015)认为Chen等(2010b)发表的新种九龙山杜鹃与大果杜鹃相同,从系统发生树看,大果杜鹃与九龙山杜鹃聚成一小支,支持率达100%,支持大果杜鹃与九龙山杜鹃为同一个种的推测。张长芹等(2015)认为百里杜鹃的一些新种的分类地位有待商榷,从黄坪杜鹃、金波杜鹃、枇杷叶杜鹃、匙叶杜鹃等形态特征推测这些新种可能是马缨杜鹃与露珠杜鹃、大白杜鹃或皱叶杜鹃的天然杂交后代; 从系统发生树可以看出,金波杜鹃与马缨杜鹃聚成一小支,支持率100%,说明金波杜鹃与马缨杜鹃有很近的亲缘关系,普底杜鹃与皱叶杜鹃聚成一个分支,同样表明二者有很近的亲缘关系。虽然不能由此明确金波杜鹃和普底杜鹃是否为杂交后代,但至少印证了二者分别与马缨杜鹃和皱叶杜鹃有亲缘关系的推测。另外,用RAD序列得到的SNP仍然无法区分某些更小分类单元的物种,比如,有鳞大花杜鹃亚组的树枫杜鹃和百合花杜鹃,以及银叶杜鹃亚组的银叶杜鹃和云锦杜鹃亚组的美容杜鹃等。目前,比较认可的观点是通过分子标记区分物种间的亲缘关系本质上与形态判断同等重要,不能顾此失彼。对于一些形态差异明显但系统进化分析无差别的种类,本文不做定论。期待后续通过更多、更准确的分子标记(比如全基因组测序技术),寻找这些近缘种在整个基因组水平的位点与结构变异; 用更科学的系统进化树构建方法进一步区分和验证。
4 结论
采用RAD-seq技术对百里杜鹃保护区杜鹃花属34个种进行测序,通过denovo组装并获得高质量SNP位点,基于这些SNP位点对杜鹃花属植物进行群体结构分析、主成分分析与系统树构建,结果表明RAD-seq技术产生的大量SNP位点可在亚属水平和物种水平区分百里杜鹃的大部分杜鹃花属植物,同时证实RAD-seq技术在复杂植物类群的物种分类方面相比传统分子标记具有明显优势。杜鹃花属6个亚属中的常绿杜鹃亚属、马银花亚属、羊踯躅亚属和映山红亚属能明显分开,而糙叶杜鹃亚属与杜鹃亚属不能区分开,表明二者亲缘关系较近。对百里杜鹃保护区新发表的金波杜鹃和普底杜鹃的分类定位进行探讨,表明金波杜鹃与马缨杜鹃、普底杜鹃与皱叶杜鹃有较近的亲缘关系,但还需进一步证实。本研究结果可为复杂植物类群的物种界定在方法上提供参考,也可为百里杜鹃保护区杜鹃花的资源开发与新品种培育提供指导。
猜你喜欢
作物学报(2022年2期)2022-11-06
社会科学战线(2022年7期)2022-08-26
中国合理用药探索(2022年7期)2022-08-16
实用心脑肺血管病杂志(2022年2期)2022-02-22
临床医学工程(2021年12期)2022-01-07
心声歌刊(2020年4期)2020-09-07
心电与循环(2020年1期)2020-02-27
北方文学(2019年21期)2019-08-18
小学生作文(低年级适用)(2019年4期)2019-04-29
小学生作文(中高年级适用)(2018年5期)2018-06-11
