
我国医疗器械主文档登记制度研究
2021-04-08 09:38赵鹏张书培史新立
中国医疗器械杂志 2021年2期
赵鹏,张书培,史新立
国家药品监督管理局医疗器械技术审评中心,北京市,100081
0 引言
按照我国《医疗器械监督管理条例》[1]以及《医疗器械注册管理办法》[2],医疗器械产品需由申请人根据产品类别申请注册或备案,取得医疗器械注册证书或备案凭证后才可上市销售。申请人在提交申请时,需提交证明产品安全、有效所需的全部资料。但是,医疗器械的生产可能存在部分原材料、部件外购,或者部分生产工艺外包的情况,从而使得有些技术资料涉及外购或外包部分内容(尤其是对于设计和工艺较为复杂的医疗器械),而供应商由于技术保密原因不愿将相关内容透露给医疗器械申请人,由此会面临注册申请的困难。为解决这一难题,医疗器械主文档(Master File)应运而生。
医疗器械主文档是技术资料的一种,该类资料由其所有者(如原材料供应商)直接提交给医疗器械监管机构,用于授权医疗器械产品申请人在申报医疗器械注册等事项时使用。一般来讲,主文档由其所有者自愿提交给监管机构进行登记,但在登记时不经过实质审评。当医疗器械申请人在申报产品上市注册时需要使用主文档资料时,主文档所有者向医疗器械申请人出具授权书。医疗器械申请人将该授权书作为申报资料的一部分以代替经登记的主文档资料。当监管机构对医疗器械申报资料进行审评时,可依据授权书调阅经登记的主文档资料进行审评。
1 美国、欧盟、日本医疗器械主文档制度介绍
1.1 美国医疗器械主文档制度
美国食品和药品管理局(FDA)对药品[3-7]、生物制品[8]、医疗器械[9-10]、兽药[11]以及烟草[12]的主文档都有相对独立的法规要求。FDA器械与放射健康中心(CDRH)于1997年2月发布了相关指导性文件[10]来规范医疗器械主文档(master files for devices,MAFs)的使用和管理。目前FDA官网上的医疗器械主文档指导性文件于2017年11月更新[9],主要更新内容是在原有的两个用途“为帮助保护医疗器械行业的商业秘密”和“促进医疗器械的合理科学评估”的基础上,增加了一个新用途“多个申请人在不同产品上采用相同材料或工艺时的,申报时也可考虑采用主文档”。可见FDA在该制度约20年的实践中也是在不断总结和更新,其不仅达到了初始设置的目的,还获得了额外的收益。
1.2 日本医疗器械主文档制度
日本厚生省于2000年12月发布了《关于建立用于医疗用具的原材料数据库(医疗用具主文档制度)的通知》,当时医疗器械主文档的适用对象仅限于聚氯乙烯、聚乙烯和聚丙烯三种原材料。2014年,厚生省把药品和医疗器械等的主文档制度统一到《药机法》第80条第6项(1)的管理范围内,并发布了《原料药等登记原簿引用指南》[13-15],规定其制定目的是为了在药品、医疗器械及再生医疗产品等的上市许可审评中,引用非申请人持有的原料、添加剂、包装材料、生产方法、非临床试验数据等审评中必要的信息,保护其知识产权,同时提高审评效率且保持稳定的质量。日本主文档制度最近的一次更新是2019年。厚生省发布了《关于医疗器械原材料主文档的管理办法》[16],在医疗器械方面主要是对适用对象范围、登记内容的记述方式等有所调整。适用对象由原先限定的聚氯乙烯、聚乙烯、聚丙烯,扩大到所有的医疗器械原材料,另外对原材料主文档的资料要求做了进一步规范。
1.3 欧盟医疗器械主文档制度
欧盟在其指令 2001/83/EC[17]中记录了关于药品主文档(European drug master file,EDMF)的相关要求,包括主文档的目的、适用范围等,后于2005年发布关于药物活性物质主文档程序的指南,2018年12月更新到第四版[18-19]。由于欧盟上市的医疗器械由各EC公告机构进行审评,形式较为灵活,因此未做关于医疗器械主文档的统一规定。
2 主文档登记制度对于行业发展的意义
2.1 促进供应商如医用原材料相关产业的发展
以原材料供应商为例:一方面,通过对原材料主文档的审评,更加规范原材料供应商的研发、生产行为,提高其研发设计能力和质量管理水平;另一方面,主文档制度的建立也为新的原材料生产企业提供一个展现其可信度的平台。通过统一的平台和审评要求可以为医用原材料生产企业创造一个良性的发展环境。
2.2 使医疗器械企业的外购和外包更加顺畅
由于社会分工的程度不断提高,很少有企业从原材料的制造到终产品的生产全部包揽,因此目前绝大部分医疗器械企业涉及外购和外包。这些外购和外包内容甚至是影响最终产品质量的关键因素。但是,能够提供优质物料或服务内容的供应商可能会因为技术秘密等原因,一般不愿配合规模较小的医疗器械生产企业提供相关资料,从而造成合作不畅,而主文档制度可以解决这个问题,从而消除医疗器械生产企业(尤其是初创型、小微企业)与大的供应商之间的“隔阂”,使之较为顺畅地实施外购和外包行为,从而保障医疗器械的质量。
2.3 为医疗器械企业的注册和风险控制带来便利
主文档制度建立后,由于存在多个医疗器械申请人在不同产品上采用一份主文档的情况,因此既可以避免企业的重复工作,也可以简化器械的注册申报,减轻企业研发注册的负担。尤其是对于在已建立主文档制度的国家和地区登记过或引用过主文档的企业,可以做到在不同国家注册申报工作间的无缝衔接。此外,在主文档制度下,若主文档的内容有变化,如外购原材料工艺变化,供应商有义务向医疗器械申请人(注册人)报告,以便由其评估这些变化对医疗器械的影响,以确定在质量体系和注册申报上是否采取相应措施,这将提高医疗器械申请人对医疗器械的风险把控能力。
3 主文档登记制度对于器械监管的意义
3.1 提高审评审批效率,减轻审评负担
建立主文档制度后,当多个申请人在不同产品上采用相同材料或工艺时,所引用的可能是同一份主文档资料。当该主文档第一次被引用并关联审评后,将会在系统中有所标记。后续不同的审评员在审评引用该主文档的产品时则会考虑其已经审评的因素,避免重复审评,同时也有助于审评尺度的前后一致性。
3.2 提高对医疗器械风险把控程度
医疗器械技术审评需要对产品安全性和有效性进行系统评价,确保对患者的受益大于风险。主文档内容可能涉及医疗器械的原材料等关键信息或与之相关的研究数据,对于系统评价产品尤其是高风险、复杂产品的风险受益比具有极其重要的作用。基于对医疗器械全生命周期的监管原则,风险越早发现则越有利,因此主文档制度有利于从医疗器械源头进行风险把控,提高技术审评的准确程度,从而进一步确保对产品安全有效性评价的充分性。
3.3 促进医疗器械监管科学的发展
医疗器械监管科学旨在开发新的工具、标准和方法来进一步评价医疗器械的安全、有效、质量和性能[20],为监管解决前沿性、交叉性、适应性和灵活性问题。主文档制度作为监管科学涉及的一种新工具和方法,为医疗器械主要原材料、添加剂、关键元器件、特殊生产过程等极大影响医疗器械终产品质量和性能的重要因素建立独特的信息通道,也为今后建立相关数据库,以及进一步搭建应用更为广泛的大数据平台奠定基础,更将对于医疗器械监管科学资源的集约化利用,以及核心生物材料、关键共性技术相关的监管科学发展具有极其重要的战略意义。
4 我国医疗器械主文档登记制度研究
4.1 主文档登记制度研究过程
国家药品监督管理局医疗器械技术审评中心于2018成立专项工作组开始进行医疗器械主文档登记制度的研究,在调研各国医疗器械监管机构主文档制度运行经验的基础上,结合医疗器械技术审评中遇到的各种情况,起草了适于我国监管现状的医疗器械主文档登记制度相关文件,并于2019年1月面向社会公开征求意见[21]。目前已结合反馈意见对制度文件进行了进一步完善,并将相关文件上报国家药品监督管理局。由于该制度的实施需要相应的法规支持,在修订《医疗器械监督管理条例》配套规章时也将考虑相关内容。同时,为便于主文档所有者进行主文档登记和更新,医疗器械主文档登记系统将融入医疗器械注册电子申报信息(eRPS)系统中。
4.2 我国主文档登记制度的主要内容
目前制订的医疗器械主文档登记制度相关文件包括拟对外发布的文件和审评机构内部工作程序文件。其中,拟对外发布的文件为《关于医疗器械主文档登记事项的公告》及其附件,包括《医疗器械主文档登记申请表》《医疗器械主文档登记更新申请表》《医疗器械主文档登记回执》《医疗器械主文档登记相关信息》《医疗器械主文档登记资料形式要求》和《医疗器械主文档登记指导原则》。其中《医疗器械主文档登记指导原则》阐述了主文档登记的基本原则、主文档内容、主文档更新、授权的相关要求以及授权书样本等。审评机构内部工作程序文件包括《医疗器械主文档登记操作规范》等。以上文件中拟建立的医疗器械主文档登记制度主要流程,如图1所示。
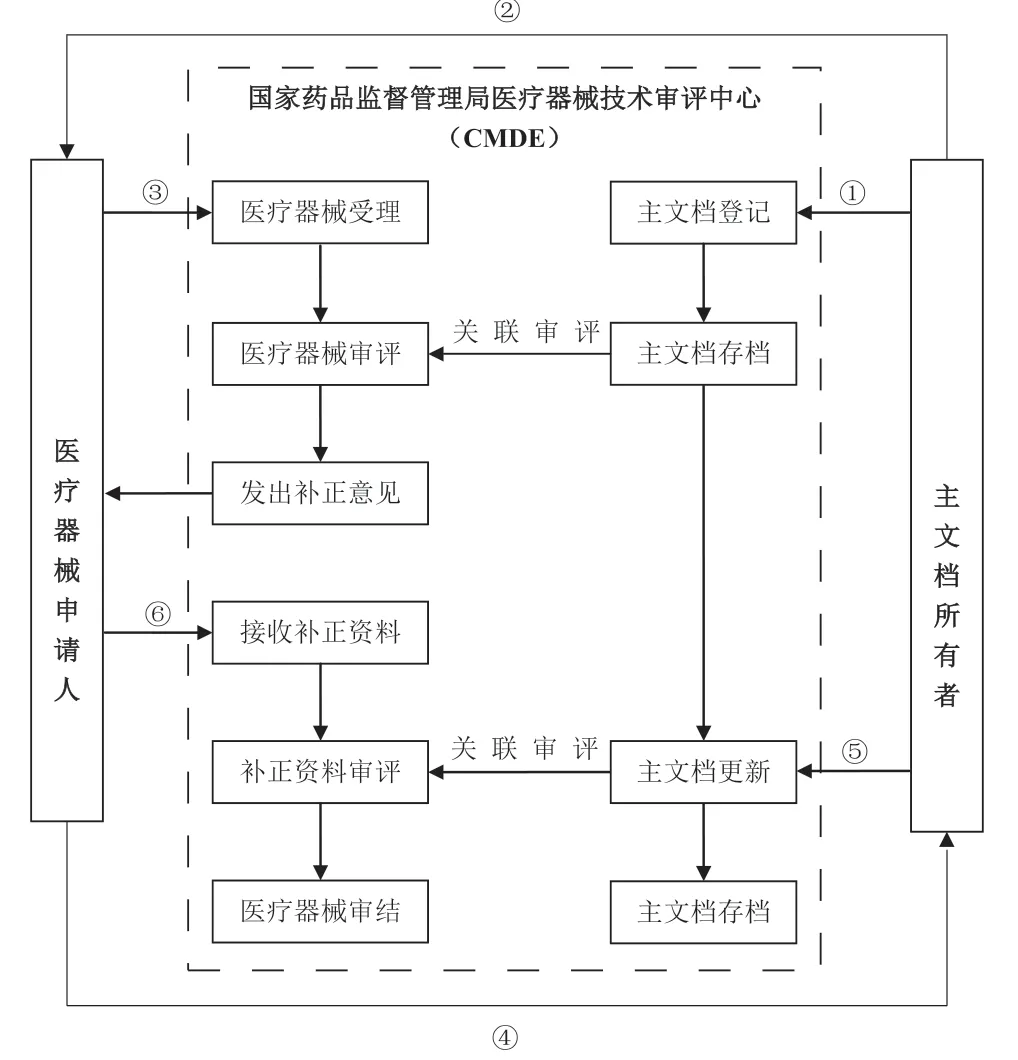
图1 拟建立的我国医疗器械主文档登记制度流程图Fig.1 Flow chart of the registration system of medical devices Master Files to be established in China
说明:
图中流程步骤按顺序分别为:
①主文档所有者申请主文档登记;
② 主文档所有者向医疗器械申请人出具主文档授权书;
③医疗器械申请人进行注册申报(申报资料中含主文档授权书);
④ 医疗器械申请人根据补正资料通知单的意见通知主文档所有者更新主文档(若补正涉及主文档);
⑤ 主文档所有者申请更新主文档(若补正涉及主文档);
⑥ 医疗器械申请人提交注册补正资料。
4.3 我国主文档登记制度的特点
虽然各个国家和地区主文档登记制度在主要目的和原则上具有共通性,但在实际操作中也有所区别。我国拟建立的医疗器械主文档登记制度相比其他国家和地区的主文档制度,具有如下特点:
(1)关于主文档应用范围:我国医疗器械主文档的内容可以应用在医疗器械外购或外包多个方面,如原材料等,并可以应用到各种医疗器械注册事项,如产品注册、变更、延续、临床试验审批等。
(2)关于主文档具体内容:医疗器械主文档的具体内容为涉及技术秘密的描述性或研究性资料,如原材料的主文档可以包括原材料组成成分描述、物理性能研究资料、化学性能研究资料、生物学评价资料/毒理学风险分析资料等。但是对于一些明确要求在医疗器械申报资料中提交的内容,并不适合采用主文档的形式提交,如体外诊断试剂原材料信息等。
(3)关于主文档内容的变化:主文档内容的实质性变化可能导致医疗器械产品的变化。因为医疗器械申请人对医疗器械的变化负责,所以医疗器械申请人需采取相应措施例如合同约定的方式使其及时知晓主文档内容的变化并进一步分析对医疗器械产品的影响。目前拟发布的医疗器械主文档登记指导原则明确医疗器械申请人可与主文档所有者以合同的形式约定主文档所有者对变化情况的告知义务。
(4)关于主文档的时效性:为保证已上市医疗器械的持续合法性,在医疗器械注册过程中直至上市后在市场上销售使用,主文档所有者对医疗器械申请人使用其主文档的授权需要始终保持有效。因此,拟发布的医疗器械主文档登记指导原则明确“一旦引用该主文档的医疗器械申请事项得到受理,则该授权不得撤销”。
5 主文档登记制度实施的相关风险分析
5.1 对医疗器械监管定位的影响
主文档制度是否会影响医疗器械监管范围和医疗器械安全有效的责任主体?主文档登记制度并非行政许可事项,仅是医疗器械注册管理中的一项措施,是一种提交资料的方式改变。《医疗器械注册管理办法》(总局令第4号)[2]第六条规定,“医疗器械注册人、备案人以自己名义把产品推向市场,对产品负法律责任。”医疗器械主文档登记制度的实施将不会改变此项规定,拟上市的医疗器械终产品的安全性、有效性,仍然由医疗器械申请人(注册人)负责。主文档所有者的责任和义务由医疗器械申请人(注册人)与之以合同形式进行约定。因此,医疗器械监管对象仍然是医疗器械申请人(注册人),而不是主文档所有者。
5.2 关于泄露技术秘密的风险
主文档制度是否会增加泄露技术秘密的风险?主文档资料实质上属于医疗器械注册申报资料的一部分,只不过这部分由主文档所有者直接提交,而不是由医疗器械申请人提交。因此,对于医疗器械注册申报资料的保密原则、保密措施等均适用于主文档资料,所以并不会由于实施了主文档制度而增加泄露技术秘密的风险。
5.3 对注册申报不确定性的影响
主文档制度是否会增加注册申报的不确定性?由于主文档是注册申报资料的一部分,因此注册申报的不确定性并不是主文档这种形式带来的,而是主文档内容包含的研究工作的充分性、合理性、规范性所决定的。对于涉及外购原材料或配件的专业知识,供应商显然会比医疗器械申请人具有更深入的理解和认识,因此由供应商直接提交资料反而更有技术优势,并不会增加注册申报的不确定性。
6 总结与展望
上述研究结果表明,主文档登记制度虽然仅仅是对提交注册申报资料方式的改变或补充,但其对我国医疗器械行业及监管体系的发展影响深远。相信在不久的将来,我国将建立起具有中国特色的医疗器械主文档登记制度,将会进一步规范和加速医疗器械上游产业发展,使医疗器械企业的外购和外包更加顺畅,注册申报更加便利,同时提高医疗器械审评审批效率和对产品风险把控的程度,提高监管的科学性,从而进一步保持和促进公众健康。
猜你喜欢
商品与质量(2021年43期)2022-01-18
医疗装备(2020年10期)2020-06-13
中学时代(2019年12期)2019-11-13
意林(2019年16期)2019-09-04
质量安全与检验检测(2019年3期)2019-07-31
留学(2019年12期)2019-07-29
质量安全与检验检测(2018年6期)2018-12-28
化学分析计量(2016年4期)2016-03-14
化学分析计量(2016年1期)2016-03-14
中国洗涤用品工业(2015年6期)2015-02-28
