
CO2氧化乙苯脱氢制苯乙烯钒基氧化物催化剂研究进展
2021-04-20 10:30葛汉青殷文超杨国庆刘昭铁刘忠文
化工进展 2021年4期
葛汉青,殷文超,杨国庆,刘昭铁,刘忠文
(陕西省合成气转化重点实验室,陕西师范大学化学化工学院,陕西西安710119)
苯乙烯(styrene,ST)作为一种重要的基础有机化工原料,广泛用于合成塑料和橡胶,主要包括聚苯乙烯(polystyrene,PS)、丙烯腈-丁二烯-苯乙 烯 共 聚 物 (acrylonitrile-butadiene-styrene copolymers,ABS)、苯乙烯-丙烯腈(acrylonitrilestyrene copolymer,SAN)等树脂,是仅次于聚乙烯(polyethylene,PE)、聚氯乙烯(polyvinyl chloride,PVC)、环氧乙烷(ethylene Oxide,EO)的第四大乙烯衍生产品。此外,苯乙烯在药品、染料、农药等合成方面的应用也表现出渐趋增加的态势。
苯乙烯的工业生产工艺主要有三种[1],即乙苯(ethylbenzene,EB)直接脱氢工艺(EBDH 工艺)、环氧丙烷/苯乙烯联产工艺(PO/SM 联产工艺)和乙苯脱氢-氢选择性氧化工艺(Smart 工艺),其中全球87%以上的苯乙烯生产装置采用乙苯直接脱氢工艺。该工艺所发生的主反应如式(1)。

这是一个分子数增加的强吸热反应,乙苯转化率受热力学平衡限制,提高反应温度、降低体系压力有利于反应向正向进行。但是从动力学角度看,提高反应温度,意味着装置能耗增加。同时,反应温度过高会加剧苯乙烯聚合、乙苯(苯乙烯)裂解生成如苯、甲苯等副反应,从而降低苯乙烯选择性,进而增加苯乙烯分离能耗。因此,在实际反应过程中,往反应体系中加入过量的过热水蒸气,以达到如下目的:①加热反应原料,补充反应吸收的热量,维持反应温度;②作为稀释气,降低乙苯分压,提高乙苯转化;③消除催化剂表面积炭,延长催化剂寿命。
工业装置通常采用铁基氧化物催化剂,在过热水蒸气(615~645℃)、水油比(水与乙苯质量比)1.4~1.8、常压或负压条件下进行,苯乙烯选择性为94%~96%,收率为64%~68%,催化剂寿命一般2~3年。
水油比是影响乙苯分压和反应温度的重要因素,而水蒸气单耗则是衡量乙苯脱氢工艺先进性的重要指标之一。降低水油比,乙苯平衡转化率随之降低。在低水油比条件下,为保持高乙苯转化率,必须提高主蒸气温度。例如,水油比为1时,主蒸气温度需接近930℃,远超现有加热锅炉的能力及高温管道、设备材质的使用温度[2]。另外,低水油比会诱发活性组分氧化铁因烧结而粒径增大,使得催化剂表面积炭增加,导致催化剂稳定性变差[3-5]。
乙苯脱氢制苯乙烯已有近八十年的研究历史,在催化剂活性相、助剂及反应工艺等方面进行了大量基础和应用技术开发工作,主要目的在于解决低水油比条件下催化剂失活和反应能耗过高等问题[6-8]。但是迄今为止,乙苯直接脱氢工艺依然存在如下主要问题:①受热力学平衡限制,乙苯转化率不高;②苯乙烯选择性比较低;③过热水蒸气余热无法回收,过程能耗大;④高温高湿环境下催化剂中钾组分流失严重,催化剂活性降低;⑤低水油比条件下,催化剂性能差,寿命短。
同时,上述问题也激励着研究人员不断探索新的替代工艺,提出了如采用膜反应器[9]、分别以O2(或空气)[10-15]、N2O[16-18]、SO2[19]或CO2[20-23]为氧化剂的氧化脱氢等多种方案,其核心思路是将乙苯脱氢反应中生成的氢移除或通过耦合反应消耗掉,从而缓解乙苯脱氢的热力学平衡限制,进而降低反应能耗。现有研究结果显示,氧化脱氢方案可能是最佳替代方案。
以强氧化剂O2(或空气)为氧源进行乙苯氧化脱氢是强放热反应,反应温度较低,一般不高于500℃,且催化剂表面不易产生积炭,但乙苯易被深度氧化为CO 和CO2,显著降低了苯乙烯收率。此外,还存在O2与乙苯混合易发生爆炸等难题[10-12]。将乙苯直接脱氢与脱除氢的选择性氧化反应分步进行的Smart 工艺尽管避免了乙苯深度氧化问题,但是反应器结构复杂,整体工艺要求苛刻,并没有得到广泛的工业应用。
如果以弱氧化剂SO2或N2O 为氧源,虽然缓解了乙苯直接脱氢的热力学平衡限制,也不存在乙苯深度氧化的问题,但由于SO2或N2O 本身及还原产物(如CS2和COS等)的毒性和腐蚀性等问题,其应用也受到限制。
Bartholomew[24]研究焦炭气化过程中各种气体的氧化能力,发现其活性顺序是O2(105)>H2O(3)>CO2(1)>H2(0.003),表明CO2的反应性远低于O2,但与H2O相当,仍表现出对焦炭较强的氧化能力。若将热力学稳定且动力学惰性的CO2催化活化作为弱氧化剂利用,不仅能够克服O2氧化低碳烷烃、乙苯等烃类脱氢面临的深度氧化等难题,提高直接脱氢的热力学平衡转化率,而且可以实现温室气体CO2的资源化利用[25-26]。此外,相对SO2及N2O 来说,CO2更加绿色,安全。
因此,目前以CO2作为弱氧化剂应用于低碳烷烃、乙苯等烃类氧化脱氢及芳构化等重要工业过程已成为该领域的研究热点[26-27]。为此,下文主要从反应工艺、反应机理及催化剂等方面对近年来CO2氧化乙苯脱氢领域的研究工作,特别是钒基氧化物催化剂体系进行总结分析。
1 CO2氧化乙苯脱氢特点
热力学和模拟计算结果表明,在相同反应条件下,采用CO2替代水蒸气进行乙苯脱氢反应,苯乙烯生产能耗将从1.5×109~1.9×109cal/t 降低到1.9×108cal/t(1cal=4.1868J),而且乙苯转化率也明显提高[28-29]。这说明与乙苯直接脱氢工艺相比,CO2氧化乙苯脱氢工艺更加节能。
图1为乙苯脱氢与逆水煤气变换耦合反应在常压、不同进料组成及反应温度下乙苯平衡转化率的热力学计算结果[30]。从图中可以看出,在相同温度和相同原料气配比(EB∶CO2=EB∶N2=1∶10,体积比)的条件下,二氧化碳氛围下的乙苯平衡转化率高于氮气氛围(仅起稀释作用)下的乙苯平衡转化率。在获得相同乙苯转化率的情况下,二氧化碳氛围下所需的反应温度明显低于氮气氛围和乙苯直接脱氢(EB only)条件下的反应温度。这说明二氧化碳在乙苯脱氢反应体系中不仅具有氮气的稀释作用,还能够推动乙苯脱氢反应平衡向正向移动,从而进一步提高乙苯平衡转化率。

图1 常压下在乙苯脱氢与逆水煤气变换耦合反应中进料组成及反应温度对乙苯平衡转化率的影响[30]
反应耦合是改善受热力学和/或动力学限制的反应过程的有效途径。通过耦合两个反应,可以有效提高主反应的平衡转化率,同时也可能降低反应温度和过程能耗[31]。在CO2氧化乙苯脱氢反应体系中,通过反应耦合,逆水煤气变换反应可以消除积炭,减缓催化剂失活,从而提高催化剂稳定性。当然,在实际反应过程中,由于受到动力学或扩散影响,转化率的高低与所采用的催化剂及反应条件密切相关。
CO2氧化乙苯脱氢的主反应如式(2)所示。根据反应条件及催化剂的差异,在一定程度上伴有逆水煤气变换反应[式(3)]和逆Boudouard反应[式(4)]等副反应[32-33]。比较式(1)~式(3)可知,CO2氧化乙苯脱氢反应也可表述为乙苯直接脱氢反应和逆水煤气变换反应的耦合,即式(2)=式(1)+式(3)。

因此,CO2在乙苯脱氢反应中可能表现为以下三方面的作用[30,34]。
(1)直接氧化还原作用 CO2在催化剂表面活化产生CO 和活性氧,继而活性氧与乙苯烷基上活化的氢反应,生成水,同时乙苯转化为苯乙烯。因此,在该过程中CO2直接作为氧化剂,补充活性氧。
(2)间接氧化还原作用 首先发生乙苯直接脱氢反应,然后脱除的氢与活化的CO2发生逆水煤气变换反应,生成CO 与水。因此,其实质是通过乙苯直接脱氢和逆水煤气变换两个氧化还原反应的耦合通道,消耗直接脱氢反应中生成的氢,从而推动乙苯脱氢反应平衡向正向移动,进而提高乙苯平衡转化率。
(3)消除积炭作用 乙苯直接脱氢反应过程通常伴有催化剂表面积炭,在CO2氧化乙苯脱氢反应条件下,CO2与催化剂表面的积炭或积炭前体发生逆Boudouard 反应,能够在一定程度上消除催化剂表面的积炭,提高其稳定性。
从上述分析可以看出,CO2氧化乙苯脱氢工艺具有以下明显优势:①乙苯直接脱氢反应与逆水煤气变换反应耦合,提高乙苯转化率;②抑制深度氧化,提高苯乙烯选择性;③抑制催化剂结焦,延长催化剂使用寿命;④过程能耗低,同时实现CO2的资源化利用。
总之,CO2氧化乙苯脱氢工艺有望替代现有乙苯直接脱氢工艺,作为一条环保、节能、高效的绿色新工艺,正越来越受到人们的关注。
2 CO2氧化乙苯脱氢反应机理
从热力学计算结果可以看出,相同条件下CO2氧化乙苯脱氢比直接脱氢具有更高的乙苯平衡转化率。现有文献报道多集中在催化剂体系的筛选与优化,而对CO2氧化乙苯脱氢反应机理的研究相对较少。由于反应条件以及催化剂类型的不同,CO2可能存在多种反应途径,反应过程难以准确分析。因此,关于CO2氧化乙苯脱氢反应机理存在不同的看法,如图2所示。
一种看法认为,CO2氧化乙苯脱氢按照式(2)进行,即催化剂中的晶格氧与乙苯分子中的氢生成水,同时乙苯转化为苯乙烯,CO2在氧空位上被活化,解离出活性氧物种补充晶格氧,并释放出CO,整个反应遵循Redox 机理(氧化还原循环机理),也称为一步法反应机理。
支持这一看法的主要是具有氧化还原性的过渡金属氧化物催化体系,如铁基、钒基氧化物催化剂等。Saito等[29]采用瞬态响应法研究了钒基氧化物催化剂分别在二氧化碳氛围和氩气氛围下的乙苯脱氢反应过程。实验发现,在氩气氛围中,反应起始阶段可以明显观测到水峰的过冲现象。XPS 结果表明,反应1h 后的钒基氧化物催化剂中几乎不存在V5+。他们认为这是由于催化剂中晶格氧与乙苯反应生成了苯乙烯和水,同时V5+被还原为低价态。水峰的快速回落是催化剂中晶格氧被快速消耗所致。在二氧化碳氛围中,也观测到水的峰,但是呈现缓慢回落的趋势。XPS结果表明,与新鲜催化剂相比,反应1h 后的钒基氧化物催化剂中仍然存在V5+,不过含量有所降低。他们认为这是由于CO2能够在氧空位上被活化,解离出活性氧物种,补充了晶格氧,将部分低价态的钒物种重新氧化到高价态的结果。但是CO2的氧化能力较弱,难以保证钒物种在整个脱氢过程中都保持初始的高价态,所以催化剂会逐渐失活。他们在铁基氧化物催化剂也发现了类似的规律,但是在铬基氧化物催化剂上则不同,因为乙苯很难将Cr3+还原到更低价态。虽然活性中心的演变过程可以用氧化还原循环机理解释,但是在反应产物的量比关系上又存在矛盾。假设CO2氧化乙苯脱氢完全按照氧化还原循环机理进行,那么反应产物中苯乙烯、一氧化碳和水的摩尔比应该为1,但是在实际反应生成的产物中苯乙烯的量远远大于一氧化碳和水的量,而且一般能检测到氢气。这说明CO2氧化乙苯脱氢反应可能存在其他反应途径。

图2 两种CO2氧化乙苯脱氢反应机理[35]
因此,有人提出另外一种反应机理。他们认为CO2氧化乙苯脱氢按照式(1)和式(3)分步进行,即第一步是乙苯直接脱氢生成苯乙烯和氢气,与乙苯直接脱氢工艺相同[式(1)],第二步是CO2与第一步乙苯直接脱氢反应中脱除的氢气发生逆水煤气变换反应,生成一氧化碳和水[式(3)],亦称为耦合机理或两步法反应机理。
Nederlof 等[35]在研究逆水煤气变换反应对CO2氧化乙苯脱氢反应的影响时,进一步提出了两步法氢溢流机理。他们认为乙苯脱除的氢不是先结合为氢气,而是通过溢流作用迁移到金属氧化物催化剂上,进而发生逆水煤气变换反应。为了研究催化剂的逆水煤气变换反应活性,他们定义了RRWGS=[CO][ST-H2]/[CO2][H2]/Keq,其中Keq为反应温度下逆水煤气变换反应的平衡常数,水的浓度可以通过反应关联苯乙烯和氢气的浓度计算获得,即[ST-H2]。理论上当逆水煤气变换反应达到平衡时,RRWGS值等于1。实验结果发现,对于钾元素调变的氧化铝负载的铁基、铬基氧化物催化剂(KFe/Al2O3、KCr/Al2O3)而言,RRWGS值在反应前10h呈现逐渐下降的趋势,并随后维持在一定水平(约30%以下),钾元素调变的氧化铝负载的钒基氧化物催化剂(KV/Al2O3)在整个实验过程中却基本维持在较高水平(约80%以上),然而单纯氧化铝负载的钒基氧化物催化剂(V/Al2O3)其RRWGS值在反应初期(约2h)快速上升达到30%左右,并随着反应时间的增加,维持在该水平。如果反应体系中没有乙苯脱氢反应,则这些催化剂的RRWGS值普遍较低,并且当这些催化剂被预附着部分炭后,RRWGS值进一步降低,其中钾元素调变的碳包覆氧化铝负载的钒基氧化物催化剂(KV/coked-Al2O3)的RRWGS值仅有20%左右。这与大部分报道中与铁基、铬基氧化物催化剂相比,钒基氧化物催化剂活性较高但失活较快的实验结果基本吻合。即钒基氧化物催化剂在反应初期具有较高的RRWGS值,可以快速消耗乙苯脱除的氢,从而推动乙苯脱氢反应平衡向正向移动,从而得到较高的乙苯转化率。但是随着反应时间的增加,积炭不断增加,抑制了逆水煤气变换反应的有效进行,使得乙苯转化率迅速下降。
支持这一看法的理由主要是在反应产物中检测到氢气,且生成的氢气与苯乙烯摩尔比低于1,同时CO2的引入提高了乙苯转化率。研究人员认为这是由于CO2与乙苯直接脱氢反应中生成的氢发生逆水煤气变换反应,推动乙苯直接脱氢反应平衡向正向移动,从而提高了乙苯转化率。但是两步法反应机理又不能完全适用具有氧化还原特性的过渡金属氧化物催化体系(如钒基氧化物催化剂),难以解释催化剂中过渡金属元素的价态变化。
因此,研究人员认为,虽然CO2氧化乙苯脱氢反应的反应条件以及催化剂类型有所差异,但是在多数情况下,两种途径同时存在,很难说哪种途径占主导地位。Sun等[30]对Fe2O3/Al2O3催化CO2氧化乙苯脱氢反应过程进行了研究,发现其两步反应途径的比例大于一步反应途径的比例。Mimura等[36]将一氧化碳来源区分为来自式(2)的一氧化碳(标记为CODH)和来自CO2与乙苯重整反应形成的一氧化碳(标记为CODC)两部分,其中CODC 依据反应副产物苯、甲苯和甲烷进行关联计算,CODH则等于总的一氧化碳减去CODC。若只存在一步法反应途径时,CODH 与苯乙烯比值应为1;若只存在两步法反应途径时,CODH与苯乙烯比值应为一条通过零点的曲线,并且随着苯乙烯收率增加而增加。实验结果发现在Fe2O3/Al2O3催化剂上,CODH 与苯乙烯比值几乎呈线性增加,在苯乙烯等于0 时,CODH 与苯乙烯比值为0.45。这说明在Fe2O3/Al2O3催化剂上两种反应途径同时存在,其中55%的苯乙烯由两步法反应途径得到,而45%的苯乙烯由一步法反应途径得到。但是这种结论并不严谨,因为没有考虑在逆Boudouard 反应[式(4)]中,也会产生一氧化碳。此外,热力学平衡转化率计算结果显示,CO2氧化乙苯脱氢时两步法反应途径比一步法反应途径更有利于苯乙烯的生成,即CO2直接参与的氧化还原作用对乙苯脱氢效率的改善不如乙苯脱氢和逆水煤气变换反应的耦合作用显著[21]。
无论是一步法,还是二步法反应机理,CO2的有效活化对提高乙苯转化率和改善催化剂稳定性都至关重要。因为高效活化的CO2不仅能够更好地通过氧化还原机理直接与乙苯反应,也有利于与氢气发生逆水煤气变换反应,而且还可以有效地与表面积炭发生反应,清洁催化剂表面,避免活性位点因被积炭覆盖而失活[32-33]。
综上所述,CO2氧化乙苯脱氢反应中一步法和两步法反应途径可能同时存在,但是哪种反应途径占据主导地位还需要结合具体催化剂和反应条件具体分析。因此,在实际反应过程中,CO2氧化乙苯脱氢反应在不同温度下的乙苯平衡转化率曲线应该介于一步法反应途径曲线和两步法反应途径曲线之间。而两种反应途径在具有氧化还原特性的催化剂上存在互相竞争,因此选择合适的载体和具有丰富氧空位的活性组分非常关键。此外,通过优化催化剂和反应条件,促进反应尽可能以两步法途径进行,有望进一步提高乙苯转化率。
3 CO2氧化乙苯脱氢催化剂
虽然CO2氧化乙苯脱氢工艺具备低温、节能、高效及环保等优点,但是从现有研究结果来看,与工业上乙苯直接脱氢催化剂相比,目前所报道的CO2氧化乙苯脱氢催化剂的综合性能还存在一定差距,主要体现在乙苯转化率低、失活快等方面。因此,研究人员针对CO2氧化乙苯脱氢催化剂进行了大量研究,主要集中在铁基[37-41]、铬基[42-43]、钒基[44-59]氧化物催化剂,还有部分涉及铈、锆、钴等氧化物及碳材料等催化剂[60-71]。
工业上乙苯直接脱氢工艺中所使用的铁基氧化物催化剂在CO2氧化乙苯脱氢反应中活性不高,因此近些年开发了如尖金石类[72-73]、合金类[37]及碳化处理[40-41]的铁基氧化物催化剂,也有部分工作是关于控制Fe2O3晶面取向[38]、采用不同形貌载体负载Fe2O3[39]等。但总的来说,催化剂活性不高,失活严重,而且铁与碳能形成多种复杂晶型的FexCy化合物,研究难度较大。铬基氧化物催化剂主要存在环境污染问题,研究工作不多。
相比铁基和铬基氧化物催化剂而言,钒基氧化物催化剂活性相对要高,并且没有环境因素的限制。因此,大量研究工作集中在钒基氧化物催化剂。已有多个研究小组对其进行了长期系统性的研究,其中部分工作已经步入中试研究阶段。因此,如下主要从钒基氧化物催化剂的活性钒物种、氧化还原性能、表面酸碱性、催化剂表面积炭及其作用等几方面总结近些年关于CO2氧化乙苯脱氢制苯乙烯钒基氧化物催化剂的研究进展。
3.1 钒物种含量及聚集形态
Fan 等[56]采用周期性密度泛函理论对V2O5(001)进行了研究,发现催化剂表面具有三种形式的晶格氧,分别是钒酸氧、桥氧和三配位氧,对应钒物种的单聚态、寡聚态和多聚态三种聚集状态。Liu 等[74]采用原子层沉积法(atomic layer deposition,ALD)制备了VOx/γ-Al2O3催化剂(图3)。结果表明,经一次ALD 循环,催化剂表面只检测到单聚态钒氧化物。当ALD循环周期数增加到3~8时,形成了聚合态钒氧化物,并且钒氧化物的聚合程度逐渐增加。而当ALD 循环周期数增加到12 时,除了聚合态钒物种外,还形成了结晶态的V2O5。ALD制备的VOx/γ-Al2O3催化CO2氧化乙苯脱氢反应的性能评价结果表明,单聚态的钒物种比聚合态钒物种以及结晶态V2O5具有更好的活性和稳定性。通过关联分析催化剂活性与结构表征结果,得出V—O—Al键是CO2氧化乙苯脱氢的主要活性位点。此外,催化剂稳定性分析结果表明,不同结构的钒物种组成,其V5+被深度还原到V3+的程度是不同的,其中单聚态钒物种的V5+被深度还原到V3+的程度最低(主要还原到V4+),因此具有最好的稳定性。伴随着钒氧化物的聚合程度或尺寸的增加以及结晶态V2O5的出现,V5+被深度还原到V3+的程度逐渐增加,进而导致稳定性逐渐变差。这与Wang 等[21]的研究结果是基本一致的。他们认为当钒负载量高于1.5mmol/g时,钒物种聚合程度较高,CO2的氧化能力不足以补充消耗的晶格氧,以维持钒物种的高价态,导致V5+被深度还原到V3+,从而造成催化剂的活性下降。

图3 原子层沉积法制备VOx/Al2O3催化剂钒物种聚集态[74]
众多研究结果表明,钒基氧化物催化剂中脱氢活性物种为V5+,催化剂活性与V5+在催化剂表面的含量、聚集形态等息息相关。钒物种的聚集形态除与钒负载量相关外,在很大程度上也取决于载体的性质。一般来说,高比表面积的载体有助于钒物种的分散。表1 汇总了钒基氧化物催化CO2氧化乙苯脱氢的代表性文献报道结果。分析表1可知,在保持钒负载量相同的情况下,HMS[54]、MCF[59]等高比表面积载体负载钒催化剂通常比ZrO2-Al2O3[57]、SiO2[75]等低比表面积载体催化剂表现出更高的活性,同时载体的织构特点以及催化剂制备方法也会对催化性能产生显著影响。
随钒负载量的增大,绝大多数催化剂的活性呈现出先增加后降低的规律。这是因为随着钒负载量增加,活性钒物种在载体表面的聚集形态在逐渐变化,从单聚态、寡聚态向多聚态过渡,最终出现结晶态V2O5。这与Liu 等[74]采用原子层沉积法建立的VOx/γ-Al2O3模型化催化剂的研究结果是一致的。如朱忖等[50]在研究VOx/nano-Al2O3时发现,当钒负载量从0.8mmol/g增加到1.2mmol/g时,催化活性降低。通过XRD 表征发现了结晶态V2O5的特征衍射峰,这说明VOx的结构已经从多聚态开始转向结晶态。Li等[59]在研究VOx/MCF时发现,当钒负载量从1.2mmol/g增加到1.6mmol/g时,也具有同样现象。
在不出现结晶态V2O5的前提下,随着载体比表面积的增大,可负载的钒含量会逐步增加,催化剂活性也相应会有所提高。但钒结构不仅与载体比表面积相关,制备方法对其影响也较大。
Li 等[58]采用浸渍法制备了VOx/Al-MCM-41 催化剂,发现当钒负载量从1.0mmol/g 增加到1.5mmol/g 时,形成了结晶态V2O5,乙苯转化率低于采用溶剂蒸发诱导自组装法制备的V2O5-Ce0.5Zr0.5O2-Al2O3催化剂[77]。Elfadly 等[55]采用微波辅助法制备了VOx/γ-Al2O3催化剂,发现微波辅助法让钒物种主要分布在载体外表面,沉淀法则主要分布在载体孔道内壁。相对微波辅助法而言,沉淀法会使得原始晶粒更加聚集,载体中微孔体积和表面积减少,纳米颗粒更加致密化。这与Betiha 等[54]的研究结果也是基本吻合的,说明微波辅助法有助于获得低聚态的VOx,同时能够抑制结晶态V2O5的形成。

表1 不同催化剂的CO2氧化乙苯脱氢反应性能
载体材料其他性质(如孔容、孔径等)、载体与钒物种之间的相互作用等也会对钒基催化剂性能产生影响。如纤维状nano-Al2O3,其优良的织构性能和网状纳米结构有利于物质的传递扩散,提高了活性物种的可接近性[50]。采用纤维状nano-Al2O3为载体时,催化剂失活现象有所减缓,抑制了积炭的生成[52]。而SBA-16 载体通畅的三维孔道结构有利于钒物种的高度分散和反应介质的扩散,使得反应介质易于接近反应活性位点,从而提高反应性能。
Kainthla等[53]比较了V2O5分别负载于TiO2-Al2O3和Al2O3载体上的催化剂性能,发现TiO2的加入提高了钒物种在载体中的分散度,改善了钒物种与载体之间的相互作用,进而提高了催化剂稳定性。同样,Zhang 等[57]用氧化锆改性氧化铝载体,发现引入的ZrO2也能够有效地改善活性物种VOx的分散性,进而提高了其催化性能。
另外,通过比对大量文献报道数据(见表1),发现了一个有意思的现象。在氧化铝载体上单钒酸盐和多钒酸盐的理论单层密度分别为2.3Vatom/nm2和7.5Vatom/nm2。以γ-Al2O3(比表面积240m2/g)初步估算,多钒酸盐的理论最大钒含量为3.0mmol/g[78-79]。在载体比表面积相当时,采用原子层沉积法制备的最优化催化剂的钒负载量为1.6mmol/g,高于其他常规制备方法。采用常规制备方法,钒负载量的优化区间一般在0.8~1.2mmol/g。Zhang等[57]报道的钒负载量达到4.0mmol/g,但是催化剂性能较差,而且从表征结果中可以看出催化剂中形成了AlVO4。
因此,催化剂性能与活性物种VOx的含量、聚集形态有关,而载体的织构性质(比表面积、孔容、孔径等)及制备方法能够显著影响VOx的含量和聚集形态。因此,采用有效的制备方法,在保持VOx处于较低聚集状态且不出现结晶态V2O5的前提下,尽可能提高钒负载量,将有助于提高催化剂活性。
3.2 催化剂氧化还原性能
钒基氧化物催化CO2氧化乙苯脱氢反应的一般规律是乙苯初始转化率很高,但随着反应时间增加,乙苯转化率逐渐降低。这是因为在反应初期,催化剂表面晶格氧浓度最高,乙苯氧化脱氢反应速率最快,但随着反应时间增加,晶格氧逐步被消耗,因此反应速率逐步降低。此外,弱氧化剂CO2十分稳定,难以高效活化,虽然能够在一定程度上被活化并解离出活性氧对晶格氧进行补充,但是CO2解离活性氧的反应速率较慢,与乙苯脱氢反应速率不匹配,无法完全弥补晶格氧的消耗,进而导致催化剂活性逐步降低,并且随着反应时间的增加,催化剂不可避免地进入快速失活阶段,同时催化剂表面积炭速率也会进一步增加,迅速覆盖了催化剂活性位点,进一步降低催化剂活性。因此,调控催化剂的氧化还原能力,提高对CO2的活化效率,是获得高性能钒基催化剂的关键。
Fan等[56]采用密度泛函计算研究V2O5(001)活性位时发现乙苯优先物理吸附于2 号氧位(桥氧,V—O—V)上,氢主要以水的形式脱除,带走晶格氧。相反,氢若以氢气的形式脱除,需要克服更大的能垒。而弱氧化剂CO2重新氧化催化剂表面具有缺陷的V2O5(001)需要3.16eV 的活化能,因此晶格氧无法及时得到补充,进而导致催化剂活性降低乃至完全失活。
Park 等[80-86]围绕VSbOx/Al2O3催化剂进行了系统性研究,并且成功推进到中试研究。锑在贫锑VSbOx/Al2O3催化剂中的作用可以用氧溢流作用解释[87]。他们发现向可还原性氧化物(“溢流受氧体”)中添加“溢流供氧体”(如氧化锑),形成的溢流氧可以高效地氧化被还原为低价态的活性物种,抑制高价态活性物种的深度还原,让氧化物表面活性物种始终维持在高价态,可以获得最佳催化性能[88-89]。
对于钒基氧化物催化剂而言,催化剂活性与表面V5+的含量相关,而V3+的形成是催化剂失活的关键原因之一。因此,保持V5+-V4+之间的快速平稳的氧化还原循环,同时抑制V5+的深度还原,对于提高催化剂活性及稳定性至关重要。锑的引入不仅作为溢流供氧体,抑制了V5+的深度还原,而且增加了活性VOx物种的分散性,提高了表面V5+的含量。同时,锑的加入降低了结焦速率,抑制了积炭的形成[80]。此外,催化剂的稳定性与催化剂中V/Sb的比值密切相关。
Liu 等[74-75,90-94]就CO2氧化乙苯脱氢钒基催化剂进行了大量研究,针对VOx/Al2O3催化剂快速失活的问题,提出了利用Ce3+-Ce4+氧化还原循环抑制V5+深度还原的催化剂设计思路,如图4所示。

图4 V2O5/CexZr1-xO2/Al2O3催化CO2氧化乙苯脱氢反应机理[90]
实验结果表明,与现有文献报道的催化剂结果相比,开发的V2O5/Ce1-xZrxO2/Al2O3催化剂体系具有较高的乙苯转化率和良好的苯乙烯选择性。反应50h催化剂依然能够保持较好的稳定性,没有发现明显失活,但是随着反应时间的进一步增加,130h后仍然存在活性明显降低的问题。究其原因,主要是由于催化剂对CO2活化能力不足,无法完全弥补其晶格氧的消耗,因此Ce3+-Ce4+的氧化还原循环反应速率与V5+-V4+的氧化还原循环反应速率不能很好的匹配,进而导致V5+深度还原到V3+,失去氧化脱氢活性。其中锆元素的引入不仅增强了铈氧化物的热稳定性能,而且也有利于形成更多的缺陷,增强了铈氧化物的氧化还原能力。随着掺杂到CeO2中Zr4+含量的增加,固溶体中的氧空位逐渐增加。从Raman光谱和XPS结果可知,Ce1-xZrxO2固溶体中随着x从0 升至0.5,氧缺陷浓度(I600/I457)、表面Ce3+的相对含量或者活性氧物种(Oβ)的数量都是逐渐增大的。Ce3+-Ce4+的氧化还原循环速率对反应活性起着重要作用,即更快的Ce3+-Ce4+的氧化还原循环速率有利于获得更高的反应活性。通过对制备方法的研究,发现当铈锆固溶体具有较大的比表面积时,催化剂具有较高的反应活性,这可能是由于比表面积较大时,铈锆固溶体颗粒尺寸较小,导致催化剂中氧缺陷浓度较高,进而有效提高了Ce3+-Ce4+的氧化还原循环速率。此外,在CO2脉冲实验中发现,无论是V2O5-Al2O3还是V2O5-Ce0.5Zr0.5O2-Al2O3催化剂,经H2还原后脉冲通入CO2,质谱中均能检测到CO的信号,V2O5-Al2O3和V2O5-Ce0.5Zr0.5O2-Al2O3催化剂上累计生成的CO量分别为33μmol/g和119μmol/g,这说明还原后的催化剂能够被CO2氧化(图5)。更重要的是,催化剂活性变化顺序(乙苯初始转化率或TOF 值)与生成的CO 总量变化顺序相匹配,表明催化剂活性与其活化CO2的能力密切相关。但是,催化剂中Ce3+的含量与CO2脉冲后释放的CO 量不成正比,说明CO2不能将催化剂中的Ce3+完全氧化为Ce4+。而CO2对Ce3+氧化的难易程度对催化剂稳定性起着关键作用。
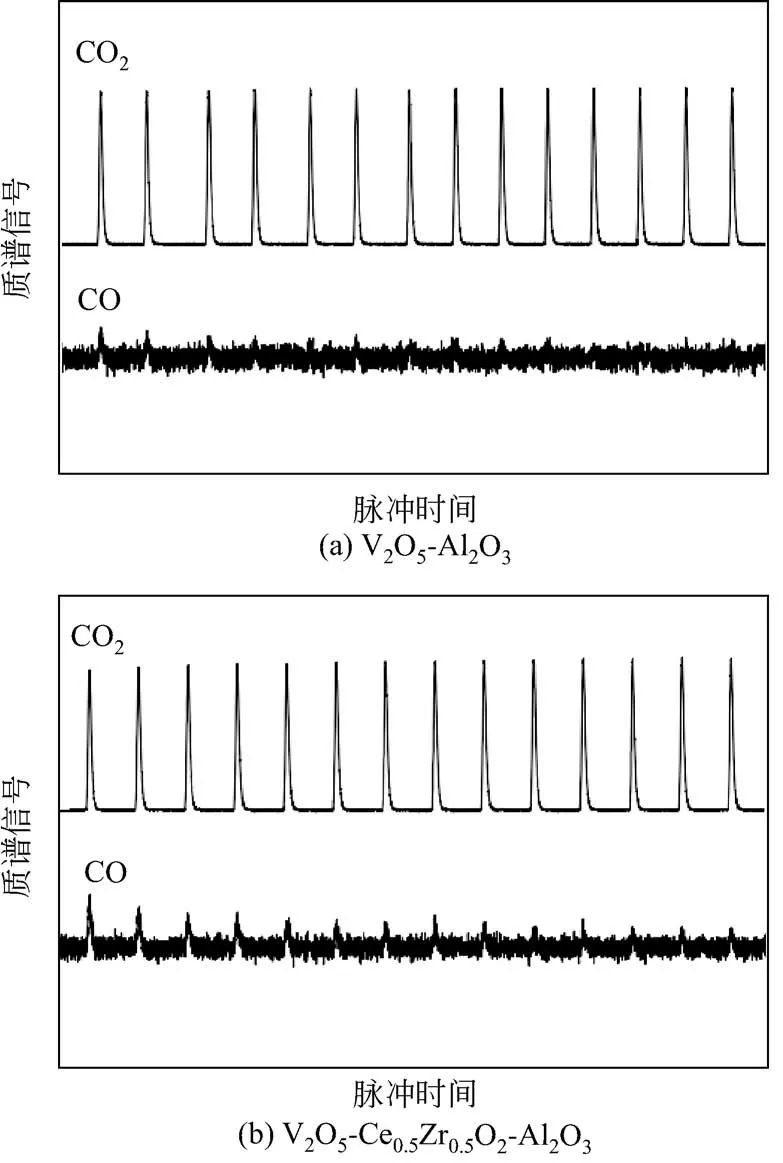
图5 经H2还原后催化剂V2O5-Al2O3和V2O5-Ce0.5Zr0.5O2-Al2O3的CO2脉冲质谱图[75]
V5+深度还原为V3+是钒基氧化物催化剂失活的关键原因之一。考虑到CO2氧化能力较弱,很多研究工作都通过引入其他具有氧化还原能力的组分,以提高催化剂的氧化还原能力,同时增强对CO2的活化,进而抑制V5+的深度还原[95-96]。采用上述策略虽然一定程度上提高了催化剂的稳定性,但是仍旧由于CO2的相对惰性使得两个氧化还原循环对的反应速率很难匹配,往往运行一段时间后,催化剂仍存在快速失活的问题。因此,针对抑制V5+的深度还原的问题,研究重点应该放在如何提高CO2氧化还原循环反应速率,使之尽可能匹配上V5+-V4+的氧化还原循环反应速率上。
3.3 催化剂表面酸碱性
除了前述活性钒物种的含量、聚集形态、催化剂氧化还原能力外,催化剂表面酸碱性也对乙苯脱氢反应性能影响较大。
Sato等[97]在研究CO2氧化乙苯脱氢反应机理时,将CO2氧化乙苯脱氢反应历程描述为乙苯首先吸附在催化剂酸性中心上,形成过渡态[C6H5—HCδ-—CH3],而乙苯中脱除的α-H与相邻碱性位上活化的CO2反应,形成过渡态[HO-C==O]δ+,然后[HO—C==O]δ+进攻[C6H5—HCδ-—CH3]中的β-H,进而脱除β-H生成苯乙烯,而[HO—C==O]δ+则形成[H2O…C==O],随后发生解离,生成H2O 和CO,重新暴露出碱性活性位。
由此可见,催化剂的酸性位和碱性位都是反应活性中心,其中酸性位吸附、活化乙苯,弱碱位脱除乙苯的α-H,强碱位活化CO2、脱除乙苯的β-H。催化剂表面存在的酸、碱中心对反应具有协同作用。如果催化剂表面酸性太强,乙苯中α-H的脱除就越容易,则会倾向于过度解离/活化乙苯的C—C 键,形成苯或者甲苯等副产物。如果催化剂表面碱性太强,虽然能增强对CO2的吸附作用,但是容易导致CO2变成CO2-3,无法解离出活性氧物种。同时,碱性太强还可能导致乙苯中的β-H 受到进攻,促进侧链C—C 键的断裂,提高对甲苯的选择性[12,98]。此外,适当提高催化剂表面碱性有利于苯乙烯脱附。因此,调节适宜的催化剂表面酸碱性是调控苯乙烯选择性的关键因素。
Wang 等[99]研究V(0.8)-Ce(0.25)/SiO2催化剂时发现,铈助剂能够调变催化剂表面酸碱性。CO2-TPD结果显示,与V(0.8)/SiO2催化剂相比,添加铈助剂后,CO2在高温区出现了明显的代表强碱位的脱附峰,相反在低温区的弱碱性位脱附峰几乎完全消失。而强碱位的存在有利于CO2的吸附与活化。因此,铈助剂的添加显著增强了对CO2的活化作用。同时,催化剂碱性的增强也有利于苯乙烯的脱附和CO2的消碳作用。此外,适量铈助剂的添加还显著提高了钒物种的分散度,起到了抑制V5+深度还原到V3+的作用,并且提高了催化剂的还原性和再氧化性能,使V5+和V4+之间的氧化还原循环更容易进行,从而显著提高了催化剂的活性和稳定性。
Hong 等[44]在研究V0.43Sb0.57Ox/Al2O3催化剂时发现,MgO 的添加增加了催化剂表面碱性,使得苯乙烯更容易脱附,有效抑制结焦,从而提高了催化剂稳定性。但一旦MgO 含量过高,会形成钒酸镁类物质,导致CO2变成CO2-3 。此外,高含量的MgO会导致催化剂表面酸性位点过少,不利于催化剂对乙苯的吸附活化,进而降低反应活性。因此,通过优化氧化铝载体中的镁含量可以同时保持较高的催化剂活性和稳定性。Chen 等[49]研究VOx/Al-MCM-41 催化剂时发现,当MCM-41 引入铝后,形成桥羟基(Si—OH—Al),出现B 酸位,总酸量增加。当负载钒物种后,形成Si—O—V 和Al—O—V 键,L酸位也有所增加。增强催化剂酸性有利于乙苯的活化。稳定性实验表明,与6VOx/MCM-41 催化剂相比,6VOx/Al-MCM-41 催化剂具有更高的积炭量,但是稳定性却明显较好,这可能是由于积炭优先形成于Al-MCM-41,而不是直接覆盖活性组分VOx。
复合氧化物催化剂往往表现出比单一氧化物更好的反应性能,这可能是由于这些复合氧化物中容易形成酸碱双功能中心,有利于调节乙苯、CO2的吸附和苯乙烯的脱附。Li 等[100]发现,向Al2O3载体中添加TiO2可以增加载体的总酸量,同时与Al2O3相比,添加TiO2后复合载体弱酸性位增加,而强酸性位有所减少。当负载钒后,载体的强酸性位减少更为明显。在乙苯脱氢反应中,强酸性位会促使乙苯裂解,而弱酸性位的存在能够减少积炭的形成。因此,TiO2-Al2O3复合载体比Al2O3载体更加有利于提高催化剂稳定性。Hong 等[83]发现采用复合载体ZrO2-Al2O3,催化剂V0.43Sb0.57Ox/ZrO2-Al2O3的碱性比V0.43Sb0.57Ox/Al2O3明显提高,因此有利于CO2的吸附活化。
催化剂表面的弱酸性位有利于乙苯吸附,抑制乙苯裂解和积炭形成,强碱性位有利于CO2吸附、活化和苯乙烯脱附。采用合适的助剂不仅可以改善活性组分的分散性,提高催化剂氧化还原能力,还能调节催化剂表面酸碱性质,从而改善催化剂性能[101-104]。
3.4 催化剂表面积炭及其作用
在CO2氧化乙苯脱氢反应中,积炭是催化剂快速失活的重要因素之一。研究人员认为积炭主要存在两种形式,即无定形积炭和石墨化积炭。前者在二氧化碳氛围下能够发生逆Boudouard 反应,消除积炭,让活性位重新暴露出来,而后者则无法被有效清除,使得被覆盖的活性位点完全失去作用。基于这些认识,近些年随着碳材料领域的发展,在CO2氧化乙苯脱氢领域也开发了很多碳基催化剂,如碳纤维、碳纳米管、纳米金刚石、石墨烯、多壁碳纳米管负载金属催化剂,且反应温度一般低于500℃。
一般而言,在反应过程中积炭会优先在高活性和非选择性位点形成,而随着积炭中C/H比例逐步增加,积炭的石墨化程度随之增加,最终会转变为不可逆积炭。由于积炭能够在一定程度上覆盖非选择性位点,因此积炭虽然降低了催化剂活性,但通常会提高选择性。因此,调控催化剂表面酸碱性,采用适量积炭包覆高活性和非选择性位点,抑制乙苯裂解,将有利于提高催化剂稳定性和苯乙烯选择性。
综上所述,活性钒物种的含量、聚集形态、氧化还原能力、表面酸碱性、催化剂制备方法等诸多因素会显著影响钒基氧化物催化剂活性及选择性。因此,采用合适的制备方法和策略,提高活性钒物种的分散度,调控催化剂氧化还原性能,维持高效的V5+-V4+氧化还原循环,抑制V5+深度还原到V3+,同时协调好催化剂表面酸碱性,增强对乙苯和CO2的吸附活化能力以及苯乙烯的脱附能力,抑制积炭的形成,是改善催化剂性能的重要关注点。
4 结语
实现CO2氧化乙苯脱氢工艺替代现有乙苯直接脱氢工艺是一个充满诱惑力,也是一个富有挑战性的课题,研究人员正在为此付出不懈的努力。鼓舞人心的是,Park 等[26]开发了一种CO2氧化乙苯脱氢制苯乙烯的新工艺,已经分别在SABIC 公司(Saudi Basic Industry Corporation)和三星通用化学有限公司(Samsung General Chemicals Co., Ltd.,SGC)实施中试研究,中试装置每天可以生产100 kg 苯乙烯,工艺流程如图6 所示。以年产60 万吨苯乙烯装置测算,每年可节省270万美元能源费用(与传统乙苯直接脱氢工艺相比,该工艺可节省33%的能源)。
提高钒基氧化物催化剂的性能,特别是稳定性,关键在于如何高效活化CO2,使之与V5+-V4+的氧化还原循环相匹配,并抑制V5+的深度还原。因此,采用原位红外等现代催化研究的原位技术,同时结合传统动态和稳态研究方法,揭示钒物种的聚集态结构、催化剂表面积炭及其结构演化等与钒基氧化物催化性能之间的关系,明确活性中心的结构及相应的反应机理,仍将是获得高性能钒基氧化物催化剂研究的关键,也是今后CO2氧化乙苯脱氢制苯乙烯钒基催化剂相关研究的主要发展趋势。
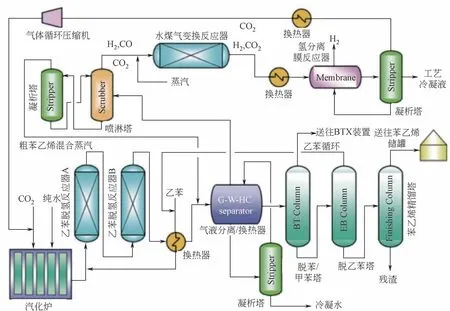
图6 CO2氧化乙苯脱氢制苯乙烯工艺流程[26]
此外,在以往研究CO2氧化乙苯脱氢过程中,对CO2的催化活化缺乏应有的重视。因此,如何同时实现CO2分子中C==O键和乙苯分子中C—H键的高效和协同活化,是设计CO2氧化乙苯脱氢高性能催化剂所应遵循的基本原则。这是CO2氧化乙苯脱氢制苯乙烯相关研究的难点,但也是开发高性能催化剂必须解决的关键科学问题,将成为该课题下一步研究的主要发展方向。考虑到CO2分子的物性、分子轨道以及热力学稳定性和动力学惰性等特点,结合近年来的相关研究进展,今后的研究重点在以下几个方面:①在催化剂中引入CaO、MgO等碱性氧化物对气相CO2进行富集,以提高其表面浓度;②CO2作为弱氧化剂的本质在于其分子中的C 原子比O原子更易于接受电子,因此可以在氧化物催化剂中加入易于给出电子的Pt、Co 等金属,从而提高催化剂的给电子能力;③从氧化物的氧化还原特性考虑,在钒氧化物催化剂中,引入富含氧缺陷的Ce、Ti 等金属氧化物,提高催化剂的氧化还原性能,可能有助于促进CO2的活化转化,进而提高其催化CO2氧化乙苯脱氢制苯乙烯的活性和稳定性。
对于CO2氧化乙苯脱氢制苯乙烯反应体系,尽管催化剂表面积炭不可避免,但其影响催化剂稳定性的关键可能不是取决于积炭量的多少,而是取决于积炭的结构或石墨化程度。这是因为,虽然一方面积炭会不可避免地覆盖氧化物催化剂的活性中心,导致催化剂失活。但是另一方面,反应初期催化剂的积炭主要以无定形或低石墨化程度炭为主,存在较多的C==O 双键等不饱和缺陷位,具有一定的催化活性,而高度石墨化的积炭没有催化活性。因此,如何抑制或延缓积炭的石墨化进程,是提高催化剂稳定性的关键。考虑到苯乙烯分子易于进一步反应的特点,采取调节催化剂的孔尺寸和结构、对催化剂表面进行亲/憎水性修饰等措施,有望强化产物苯乙烯的脱附,延缓积炭的石墨化进程,进而有助于提升催化剂的抗积炭能力和稳定性。因此,建议深入研究CO2氧化乙苯脱氢催化剂的积炭组成、结构(石墨化程度、官能团等)和积炭动力学,以厘清积炭影响催化剂性能的作用机理,从而进一步指导高效钒基氧化物催化剂的设计。
另外,结合催化剂失活特点及积炭影响催化剂失活的上述认识,从反应工艺的角度看,采用移动固定床或提升管反应器等进行工艺优化也具有重要的研究价值,将有助于进一步推进CO2氧化乙苯脱氢制苯乙烯绿色过程的工业化应用。
猜你喜欢
陶瓷学报(2021年4期)2021-10-14
陶瓷学报(2021年1期)2021-04-13
陶瓷学报(2020年6期)2021-01-26
石油化工技术与经济(2020年4期)2020-12-31
化工与医药工程(2020年4期)2020-01-09
石油炼制与化工(2020年7期)2020-01-05
汽车维护与修理(2018年7期)2018-10-13
中学化学(2015年9期)2016-04-14
世界热带农业信息(2014年11期)2015-01-05
燃气轮机技术(2014年4期)2014-04-16
