
分子动力学模拟及其在微晶玻璃中的应用综述
2021-04-29 05:56李杨井敏武吉伟刘立强
山东建筑大学学报 2021年2期
李杨井敏武吉伟刘立强
(1.山东建筑大学 材料科学与工程学院,山东 济南250101;2.国家包装产品质量监督检验中心,山东 济南250100)
0 引言
微晶玻璃作为一种新型无机材料,具备优良的化学、力学、机械、光电性能,有着广阔的应用前景,相关研究一直是关注热点[1]。 传统微晶玻璃的制备通常从改变原料配比和热处理方式等方面开展。为得到一组可行有效的配方,通常需要反复的实验,不仅消耗了大量的人力、物力,而且成效低并存在产品易开裂、成品率低、结构和性能关系尚不明细等问题[2]。 随着微晶玻璃研究的深入,要求从微观和介观角度能够准确、定量地描述微晶玻璃结构以及析晶过程规律,如研究微晶玻璃高温熔体液体结构和性能的关系、碱金属氧化物添加量对高温熔体的黏度的影响、晶核剂的形核机理等。 分子动力学模拟作为凝聚态物理学常用的计算机模拟技术,从原子、分子层次认识物质的组成,利用计算机直观展示和量化晶体结构,通过构建物质的微观结构和数值模拟热力学运动过程,得到每个粒子运动规律、能量波动等信息[3-4]。 微晶玻璃体系一般性模拟集中在三元或四元体系,完全可以借助基于分子动力学开发的模拟软件开展工作,通过调整密度大小、粒子的数目、力场参数、施加的温度、压力,构建需要的微晶玻璃模型,从微观层次上对微晶玻璃进行结构优化,从而表征宏观性能,对于改进微晶玻璃的性能从而指导生产优良性能的微晶玻璃具有社会价值。
1 分子动力学模拟概述
1.1 分子动力学的发展历史
20 世纪50 年代初,ALDER 等[5]用分子动力学方法设计了一个具有周期性边界条件的粒子系统解决有关动量与压力的问题,第一次真正意义上从微观角度模拟了物质宏观性能。 20 世纪70 至80 年代,ANDERSEN[6]对传统的动力学方法加以约束条件,开创了恒压条件下分子动力学方法。 GILLAN等[7]提出非平衡状态动力学方法。 NOSE[8]提出恒温条件下分子动力学方法。 CAR[9]解决了半导体和金属势函数难以模型化的问题。 CAGIN 等[10]提出了基于巨正则系统的分子动力学方法。 这些方法的提出都极大促进了分子动力学的的发展。 进入21世纪,随着基础学科基本理论的完善、计算机硬件的更新换代以及考虑更多作用力的多体势函数的开发,使缓慢发展的分子动力学得到迅速发展,已广泛应用于材料、医药、机械、化学、生物等多个学科,包含了晶体、非晶体、液态溶液、复合结构等方面,尤其对于材料极限条件(超临界、深过冷、高温、高压)的实验,分子动力学模拟优越性显著[11-16]。
1.2 分子动力学模拟的原理
第三种科学研究手段是除理论分析和实验观察之外的分子动力学方法,也被称为“计算机实验”手段[17]。 分子动力学(Molecular Dynamics,MD)方法和蒙特卡洛(Monte Carlo,MC)方法既有相同点又有所区别,最初MD 和MC 方法的产生的原因一样,都是为了计算积分,不同点在于MD 方法在微正则系综算积分,而MC 方法在正则系综算积分;与MC 方法相比,MD 方法优势在于计算的内容和性质更多,得到的信息更多,而且MD 方法可使用的软件要远多于MC 方法,适应于大多数体系。 分子动力学模拟是借助MD 模拟软件[18](如Moldy、Materials Studio、LAMMPS、DL_POLY 等)在原子及分子水平对多体系统进行求解的计算机模拟方法,在模拟过程遵循两个假设[19]:(1) 体系中粒子运动遵循牛顿运动定律;(2) 粒子之间的相互作用满足叠加定理。忽略量子效应和多体作用的模拟研究对象,微观粒子的运动定义为体系原子核的运动,因此忽略了原子内电子的分布情况,这与实际存在的物理系统有一定的差别。
分子动力学模拟体系内的所有粒子的运动都遵循牛顿运动方程,即Fi(t)= miai(t) ,Fi(t)为粒子i在系统中受到的力;mi为粒子i的质量;ai(t)为粒子i的加速度。 势能函数U对坐标ri求导,可得对于体系任何一个粒子,都由式(1)和(2)表示为
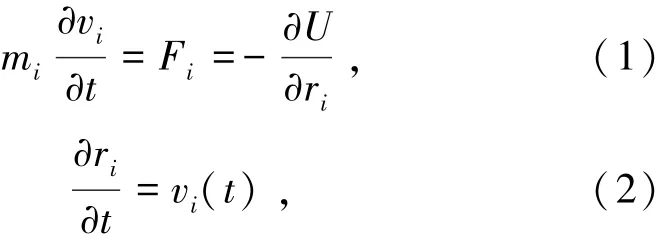
式中v为速度矢量,对式(1)和(2)求解,可以求得体系中任一粒子的位置和速度。
1.3 有限差分算法
对于多元体系,牛顿运动方程求解有一定的局限性,一般利用有限差分法完成对运动方程的求解,常用的方法包括Verlet 算法、Velocity-Verlet 算法、Beeman 算法、Gear 算法、Rahman 算法、蛙跳算法(Leap-frog algorithm)、Nordsieck 算法。
1.3.1 Verlet 算法
其算法计算过程较简单,对计算机性能要求不高,每次积分只计算一次力而且时间可逆,因此运用最为广泛,而其缺点是计算结果精度不高、轨迹与速度无关[20]。
1.3.2 Velocity-Verlet 算法
Velocity-Verlet 算法,其特点是可以同时计算出粒子一段时间内的位置、速度、加速度,并且准确性较高、计算量适中,已广泛应用于分子模拟中。 其缺点是计算过程相较Verlet 算法复杂,用时较长[21]。
1.3.3 Beeman 算法
相较于Verlet 算法,Beeman 算法更为复杂,是一种更为精确的速度表达式,其动能是由速度计算直接得到的,并尽可能得到能量守恒结果,但是实际应用中对模拟条件要求较高,而且计算量较大[22]。
1.3.4 Gear 算法
Gear 算法由泰勒式展开,能够预测每一个粒子新位置、速度、加速度[23]。
试验地位于合作市卡加曼乡新集村的甘南州农科所综合试验站,海拔2 721 m,年平均气温3.0℃,年降水量639.8 mm左右,无霜期93 d左右,耕种亚高山草甸草原土,旱川地,地力中等,前茬作物为油菜。播种时施尿素150 kg/hm2、磷酸二铵225 kg/hm2作基肥一次性施入,人工犁开沟溜种条播。2017年4月3日播种,6月6日中耕除草,田间管理略高于大田。
1.3.5 Rahman 算法
其算法表达式很复杂、计算量较大,但是计算结果精确,不适用于中小型体系模拟。 实际中很少采用Rahman 算法[24]。
1.3.6 蛙跳算法(Leap-frog algorithm)
蛙跳算法由Verlet 算法发展而来,相较Verlet式带有t时刻的速度,不计算2y(t) 和y(t - δt) ,减少了计算量,提高了精确度,轨迹与速度无关。 其缺点是位置项和速度项不能同时计算[25]。 其表达式由式(3)表示为
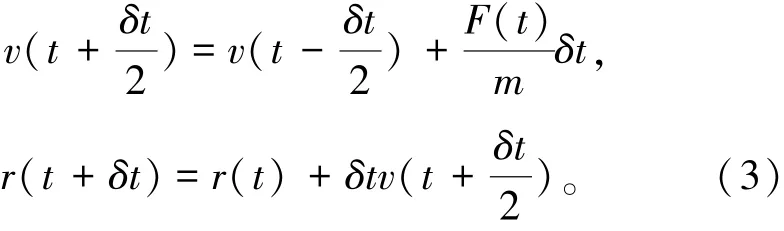
1.3.7 Nordsieck 算法
Nordsieck 算法适用于解决大系统粒子牛顿方程的求解[26]。
1.4 势函数
用来描述系统原子间或者分子间作用力的函数称为势函数[27],也称为力场。 势函数不仅决定模拟能否顺利进行,还将影响计算结果的精度。 精准且简练的势函数往往会使模拟效率提高,同时模拟结果符合物理规律。 20 世纪中后期,ALDER 等[5]借助数学方法,将粒子形象化硬球,一定距离之内会发生弹性碰撞,这种模型虽有缺陷,但是处理问题的思想促进了势函数的发展,势函数深刻体现了力场的关系。 在广大科研人员共同推动下,出现了多种形式的势函数,一般来说,根据相互作用力包含的粒子数分为二体势、三体势及多体势,目前对势、无方向性多体对泛函数、考虑角度效应多体势是最常见的种类[28]。 常见的势函数见表1。

表1 常见的势函数表
1.5 系综
2 微晶玻璃的分子动力学模拟综述
2.1 分子动力学模拟对微观网络结构的分析
微晶玻璃的制备中存在高温熔融阶段,微晶玻璃高温熔体的微观网络结构特征一直是模拟的重点问题。 需要结合均方位移函数(Mean Square Displacement, MSD)、 径 向 分 布 函 数( Radial Distribution Function,RDF)、键长键角分布、配位数和桥氧数量等多个角度进行综合分析,粒子在固定时间段内位移平方的平均值为均方位移[31]。 均方位移的量与原子的扩散系数存在着对应关系,均方位移数值越大,粒子的扩散系数越大。 对于研究微晶玻璃网络结构,由MSD 可以推测扩散系数小的粒子很可能为网络形成子或者网络中间子,反之,则为网络修饰子(不参与网络结构的构建)。 RDF 描述的是某个原子周围其他原子的分布特征,反映出给定某一粒子的位置坐标,在其半径r的空间范围内发现另一原子的概率[32],用于表征材料的有序程度,利用这一特性,可以探究微晶玻璃体系粒子之间的相互结合能力大小。 键长、键角分布规律能反映出粒子键能强弱。 配位数是原子的第一近邻原子的个数,桥氧定义为连接两个网络四面体且同时被两个四面体所共用的氧粒子[33]。 桥氧数量的多少反映了硅氧网络的完整性、致密性。 在微晶玻璃网络结构中,四面体是构成网络骨架的主要部分,与桥氧数量类似其也能反映网络的致密性[34]。
2.2 分子动力学在微晶玻璃研究中的应用进展
分子动力学模拟由于独特的实验操作环境,在国内发展较快,受到越来越多的科研人员的青睐,为了更好地了解微晶玻璃微观结构性质,指导特定性能微晶玻璃的生产,开展分子动力学模拟研究微晶玻璃的性能十分必要,但是目前在微晶玻璃领域的应用还较少。 ROSSANO 等[35]利用分子动力学模拟了CaO-FeO-2SiO2系玻璃,得到了铁周围氧原子的径向分布情况,测量了四配位和五配位铁原子的氧原子平均距离分别为1.99 和2.15 nm,这些基础的有关原子距离的模拟为以后的诸多模拟正确性评价提供了一定的参考价值。 FUMIYA[36]选取NPT 系综,采用Ewald 求和法模拟了Na2O·3SiO2熔体,发现Na—O 和Na—Na 的距离随压力的增加而缩短,Si—O—Si 键角随压力的增加而减小,说明了—Si—O—网络的崩溃,表明了—Si—O—网络的变形自由度增加,网络在高压下趋于崩溃,正由于这些结构的变化,Na2O·3SiO2熔体产生了致密化。 相较大多数模拟以单一组分对其他基础玻璃成分的影响,研究压力变化与微观网络结构的关系打开了另一种思路,基础组分固然影响宏观性能,但是某些特定温度、压力变化等外界因素也应被重视。 朱才镇等[37-38]采用了一种二体和三体的多体相互作用势探究了CaO-Al2O3-SiO2系微晶玻璃成分比例组成、微观结构之间的关系,发现Ca/Al=1/2 时,CaO-Al2O3-SiO2系微晶玻璃并不是传统理论中认为的完整的网络结构,而是存在一定的非桥氧,Ca/Al<1/2 时,Si比Al 更容易形成网络中间体。 通过模拟了不同Ca、Al 元素比情况下网络结构的特征变化,发现了桥氧的存在,这是对传统硅酸盐网络结构完整的观点的一次否定,体现了计算模拟用来解决现实问题、提供理论创新的作用。 吴永全等[39-40]采用了BMH势函数的基础上,利用经典分子动力学模拟xCaO-(1-x)Al2O3(x为成分的变化)高温熔体,重点探讨了有关Al 的配位数及其网络中的构成、微结构单元分布等结构性质,发现Al 的微观结构单元是四面体的形式,类似硅酸盐网络中Si—O 键的结构特点,证明了Al 的配位数为4,Al 在网络架构中起到了网络形成子的作用。 可以看出,大多数的模拟是以基础玻璃、三元体系为主,这是因为元素越多,需要考虑的作用力越多,开发的势函数越复杂,因此分子动力学研究领域中,开发简洁而且精确的势函数是重要的方向。 王艳伟等[41]采用EAM 原子势,在NPT 系综条件下模拟了Ni 的含量变化对Ni-Zr 非晶玻璃微观结构的影响,发现ico+other(ico 为体系的十二面结构,other 为未知的配位结构)结构随着Ni 数量的增加先增加后减少,ico+other 结构反映了非晶的形成能力,当Ni 含量为50%时,结构数量最多,即说明了一定数量的Ni 有利于促进玻璃的形成。 赵亚贤等[42]利用经典分子动力学模拟对比了Na-Al-Si系微晶玻璃和高碱高铝酸盐玻璃中碱金属的扩散行为,统计了两种玻璃的Si—O、Al—O 键长,扩散系数,发现高碱高铝酸盐玻璃中碱金属离子的扩散系数更大,这是因为[AlO4]四面体的存在,相较于[SiO4]四面体体积更大,会使网络结构疏松,有利于碱离子扩散,证明了高含量的Al 为碱金属离子的扩散提供了更多的扩散通道。 王海龙等[43]采用Mishin 嵌入原子势模拟了金属玻璃中的Cu 在急速冷条件下的形成过程,利用Verlet 算法求解运动方程,证明了随着应变率的提高,Cu 的塑性流动应力增大,晶化程度增加,应力效应和温度效应共同作用导致了金属玻璃的晶化,对于解释金属的晶化起到了一定的促进作用。 肖成[44]构建了CaO-SiO2-Al2O3-Na2O四元体系的微观玻璃模型,建立了1 873 K下体系中温度和黏度的函数关系,发现Al2O3浓度高的体系倾向于形成更加复杂的网络结构。 BINGHUI 等[45]巧妙设计了一种自上而下的模拟方法,探究影响Al2O3-SiO2系微晶玻璃断裂韧性的因素,发现并证明了增强程度随着纳米晶体的尺寸、长径比和排列角度的变化而变化。 王亚文[46]模拟了形核剂在熔融态高炉渣中的扩散行为,高炉渣成分与微晶玻璃成分相似,非均匀形核过程需要加入晶核剂降低析晶活化能。 通过数值与物理模拟发现形核剂的混匀时间与形核剂密度、等效直径成正比,与熔体黏度成反比。 可以看出,有关微晶玻璃的模拟已经取得了一些进展,从键长、键角的分布,温度、压力等外界因素对网络结构的影响,以及各元素在网络中的位置、作用等都做出了一定的解释,这些研究为制备性能优异的微晶玻璃提供了一定的帮助。
井敏课题组在微晶玻璃的制备方面经过多年的积累,已有一套成熟制备微晶玻璃的工艺。 对于微晶玻璃微观结构的探究,引进了先进的材料工作室(Materials Studio,MS)软件开展工作,取得了一系列成果。 张国莹等[47]基于MS 软件利用分子动力学模拟了Na2O-Al2O3-SiO2微晶玻璃体系熔体结构,发现了作为网络游离子的Na+能够促进Al3+从网络修饰子变成网络形成子,且在一定范围内,随着Na+的增加,Si—O—Si 和Al—O—Al 键角的分布范围变小。 王健健等[48-49]探究了Fe3+在CaO-Al2O3-SiO2系微晶玻璃中的行为,发现随着Fe3+含量的增加Al—O 键长分布更广,峰型有单峰向双峰转化的趋势。 王正[50]针对RO/R2O-Al2O3-SiO2熔体黏度特性进行了相关模拟工作,采用BMH 势,选用Shear模块计算了Na-Al-Si 系和Ca-Al-Si 系基础玻璃的剪切黏度。 发现随着碱金属元素的减少,Na-Al-Si系黏度增加要大于Ca-Al-Si 系,整体结构上,Na-Al-Si 系相较Ca-Al-Si 系网络结构较松散。 张永豪等[51]结合BMH 势,建立了MgO-Al2O3-SiO2-TiO2系微晶玻璃的微观结构模型,分别探究了TiO2晶核剂含量和SiO2含量对微观结构的影响,发现随着SiO2添加量的增大,体系中Mg2+的均方位移逐渐减小,当SiO2的含量为57.1%时,桥氧总量最多,硅酸盐网络结构最完善。 TiO2含量增加会使Al—O 键长的分布更广,峰形变宽。
3 展望
目前,采用分子动力学模拟技术对于微晶玻璃体系的模拟已经取得很大的进展。 从微观角度出发,通过原子间相互作用力、边界条件的选取、热力学性质、运动方程的计算对微晶玻璃宏观的性质(黏度、致密度、硬度等)做出了解释,改进了微晶玻璃的性能。 但是仍存在一些问题,如初始模型过于理想化,而实际晶体材料存在各种缺陷,应当深入研究构建有缺陷的建模方法;MD 方法有观测时间和系统大小的限制,仿真的规模较小,原子数较少,缺乏普遍性,应当发展较大规模的模拟;微晶玻璃通常使用经典分子动力学模拟,忽略了电子极化效应,对于电荷的相关信息无法获取,势函数过于依赖力场参数,数量较少,更加精确和简洁的势函数是发展分子动力学的重点;微晶玻璃模拟的模型也较为简单,原子种类和数量较少,大多数模拟忽略了含量稀少的元素,或许会影响宏观性能。 因此,多元体系的微晶玻璃的分子动力学模拟是发展的方向之一。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
当代陕西(2022年4期)2022-04-19
建材发展导向(2021年13期)2021-07-28
建材发展导向(2021年13期)2021-07-28
佛山陶瓷(2018年8期)2018-11-07
科学与财富(2016年15期)2016-11-24
科技视界(2016年18期)2016-11-03
诗选刊(2015年6期)2015-10-26
软科学(2014年8期)2015-01-20
对联(2011年8期)2011-09-18
