
花发育相关基因分子进化与花发育调控网络拓扑中心性的相关性研究
2021-05-11 05:53张頔高晓阳张轩刘长宁
安徽农业科学 2021年8期
张頔 高晓阳 张轩 刘长宁


摘要 探索了花发育基因调控网络的3个拓扑中心性(连接度、中间性和接近度)与其中基因分子进化速率的相关性。结果发现,随着网络中心性的增加,基因的序列将更加趋向于保守,即基因的进化速率与中心性参数呈负相关。这一结果与拟南芥蛋白质相互作用网络中所观察到的模式一致,也许是因为多效性制约了进化。
关键词 花发育基因;基因调控网络;分子进化;网络拓扑中心性
中图分类号 Q75文献标识码 A文章编号 0517-6611(2021)08-0001-04
doi:10.3969/j.issn.0517-6611.2021.08.001
开放科学(资源服务)标识码(OSID):
Correlation between Molecular Evolution of Flower Development Related Genes and the Topological Centralities of the Flower Development Regulatory Network
ZHANG Di1,2, GAO Xiao-yang1, ZHANG Xuan1,2 et al
(1.Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, Menglun, Yunnan 666303;2.University of Chinese Academy of Sciences, Beijing 100049)
Abstract In this study, we investigated the correlation between three topological centralities (connectivity, betweenness and closeness)of regulatory network of flower development, and the molecular evolutionary rate of the related genes. It was found that with the increase of network centralities, the gene sequences would tend to be more conservative, i.e., the evolutionary rate of genes was negatively correlated with the network centrality parameters. These results were consistent with the patterns observed in the Arabidopsis thaliana protein-protein interaction network, may be because pleiotropy constrains evolution.
Key words Flower development genes;Gene regulatory network;Molecular evolution;Network topological centrality
基因总是处在一个特定的生物功能网络环境中发挥作用。这些功能网络是由分子及其相互间的作用构成,如蛋白质相互作用网络是由蛋白质及其之间的相互作用构成,而基因调控网络是由转录因子、被调控的靶基因及它们之间的转录调控关系组成。目前大规模的基因组、蛋白质组和相互作用组等组学数据的涌现,为了解这些真实生物中运行的网络提供了新的机遇,同时也为分子进化研究提供了一個新的视角。从传统地关注单个基因的分子进化,转移到在整个网络中的分子进化研究时,人们通常会关注基因在网络中的拓扑属性与基因进化速率的关联,之前已有许多研究发现生物网络中反应基因中心性的拓扑属性(连接度、中间性和接近度)与进化速率有相关性。如在酵母和果蝇的蛋白质-蛋白质相互作用网络中的连接度[1-2]、中间性以及接近度[3],人类、拟南芥、水稻、番茄、葡萄和玉米的基因共表达网络中的连接度[4-5]和酵母的转录调控网络中的中间性相继被报道[6],但对花发育基因调控网络的研究相对较少。
拟南芥的花发育过程是由一个复杂精细的基因调控网络控制[7]。首先,在该网络的上游,开花时间受到多条成花途径的调控,这些途径汇聚在信号整合因子上。然后这些整合基因会激活分生组织身份基因,最后再激活花器官特征基因,进而调节不同花器官身份(如花萼、花瓣、雄蕊、心皮和胚珠)的分化过程。研究发现花发育调控网络几个阶段的基因进化速率之间有所差异[8],但在拟南芥的花器官细胞身份调控网络中,发现进化速率与所研究的拓扑属性之间无显著相关性[9]。花器官细胞身份调控网络能否代表整体花发育网络的性质,目前尚不清楚。
笔者收集和整理了拟南芥的花发育相关核心基因,并计算了其蛋白质编码序列在4个十字花科植物(Arabidopsis lyrata、Brassica oleracea、Brassica rapa和Capsella rubella)中的进化速率,进一步评估了基因的进化速率与其所在花发育调控网络中的拓扑中心性(连接度、中间性和接近度)之间的相关性。
1 材料与方法
1.1 花发育基因调控网络
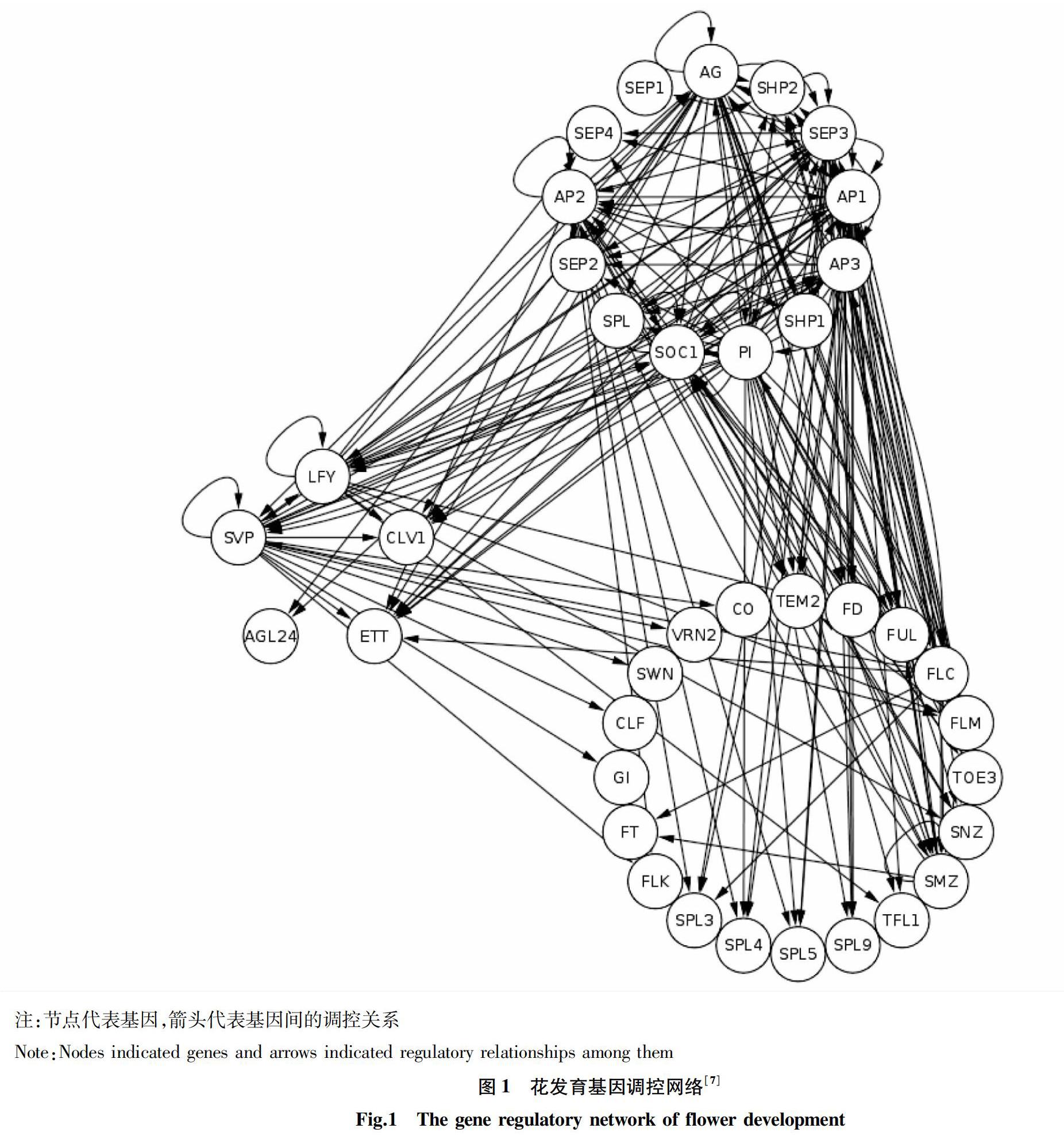
花发育基因调控网络中的数据主要来自于Pajoro等[7],对参与花发育的基因及其相互作用关系进行了综述,其中的调控关系主要是通过染色质免疫共沉淀(ChIP)试验来确定。除2个miRNA后,基因调控网络包含38个蛋白质编码基因和201个转录调控关系(图1)。借助拟南芥信息资源(TAIR)[10]中的基因功能数据信息,调控网络中的基因被分成3个主要类别。
1.2 直系同源基因的识别
十字花科的5个已测序物种(Arabidopsis thaliana、Arabidopsis lyrata、Brassica oleracea、Brassica rapa和Capsella rubella)的蛋白质和CDS(nucleotide coding sequences)序列的数据分别从Phytozome、NCBI和Ensembl Plants基因组数据库上下载。
为了找出每个拟南芥基因在其他4个物种中的直系同源基因,在拟南芥的蛋白质序列和另一个物种的蛋白质序列之间分别进行了双向最优局部比对搜索BLAST[11](使用1e-15的E-value)。结果再次进行过滤和筛查。直系同源基因过滤和筛查控制标准:比较少的缺失氨基酸,以及相似度高的基因。
1.3 网络中心性的计算
对于每个基因,使用Python软件包NetworkX[12]分别计算了连接度(degree)、中间性(betweenness)和接近度(closeness)3种拓扑网络中心性。其中,连接度是指一个节点所连接的节点数,也被称作“connectivity”;中间性是指所有最短路径通过一个节点的比例;接近度是指一个节点与所有其他节点的平均距离的倒数。
1.4 基因进化参数(dN/dS、dN和dS)的计算
首先使用MAFFT[13](--auto参数)对每一组直系同源基因的蛋白质序列进行多重序列比对。随后,根据蛋白质的比对结果,使用PAL2NAL[14]对CDS序列进行了基于密码子的多重序列比对。使用Gblocks0.91b软件[15]将比对结果中质量较差的部分进行了裁剪,使用的参数:-t=c -b4=5 -b5=h。此外,应用PhyML 2.4软件[16]中的最大似然方法进行了系统发育分析,其中配置參数用SMS[17]进行优化选择。基因的进化速率(dN/dS)、非同义替换率(dN)和同义替换率(dS)是基于上述CDS密码子比对和进化树,使用PAML 4[18]中的CODEML软件进行计算。
1.5 参数间相关性的统计分析
运用R语言3.4.4(http:∥www.r-project.org/)的环境进行有关统计分析。网络中心性(连接度、中间性和接近度)与进化相关参数(dN/dS、dN和dS)之间的相关性,采用斯皮尔曼等级相关系数(spearmans rank correlation coefficient)来进行衡量(使用了R中的Hmisc包)。如果相应零假设检验的P< 0.05,则认为被检测的网络属性与进化相关参数之间有显著相关性。在整个计算流程中,自行编写Perl和Python脚本进行数据格式调整。
2 结果与分析
2.1 网络中心性的计算结果
使用的是Pajoro等[7]的研究中所总结的花发育基因调控网络,网络中包括38个编码基因和201个互作关系。分别计算了网络中每个节点反应网络中心性的3个参数(连接度、中间性和接近度),计算结果见表1。
2.2 基因进化参数的计算结果
通过蛋白质双向最优比对和质量过滤后,得到了花发育相关基因在其他4种十字花科植物中的直系同源基因。结果显示,在研究的拟南芥38个基因中,有35个基因在4种植物中都找到了双向最优比对的基因,但其中FLM、PI和SNZ基因在某些植物中没有找到同源基因。通过对没有缺失值的35组直系同源基因使用PAML中的最大似然算法估算出了dN/dS、dN和dS。从dN对dS的比值(dN/dS)推断自然选择的影响。通常,适应性变化可以通过分子水平计算非同义替代速率dN与同义替代速率dS的比值进行分析。如果没有选择作用,或没有很强的有害突变,同义与非同义替代的速率相同,则dN/dS=1;如果存在负选择,则dN/dS<1;如果存在正选择,则dN/dS>1。因此,dN/dS不仅可以用来检测选择作用,还可以用来确定选择方向。所研究基因的dN/dS值都小于0.4(表2),平均值为0.178 8。表明这些基因总体上都是在纯化选择下进化的,只是在进化过程中受到了不同的选择约束。
2.3 进化参数与网络中心性之间的相关性分析
分别计算了基因编码区进化参数与网络中心性之间的相关性,结果见表3。经过统计检验,发现基因序列的非同义替换率(dN)与同义替换率的(dS)的比值(dN/dS)与网络中心性(连接度、中间性和接近度)呈显著负相关,这可能指示着处于网络中央的基因受到了更多的功能限制,而倾向于减少非同义替换的纯化选择。dN的相关性也反映了类似的负相关趋势。其中与中间性的负相关性最显著,与连接度的负相关性次之,而与接近度的负相关性不显著。另外在dS与网络中心性之间没有发现相关性。
该研究结果与之前在蛋白质-蛋白质相互作用网络[3,19]和共表达网络[4-5]中所得出的研究结果一致。似乎在这些分子网络中,连接度越高的基因可能会因为具有多效性,而在进化上更加保守[20];这表明由于越中心的基因序列改变会对生物体产生更有害的影响。那么为何在拟南芥的花器官细胞身份调控网络中没有发现进化速率与拓扑属性之间有显著的相关性[9],原因之一可能在于,根据Liu等[8]所述花发育基因调控网络所分成的几个功能子网,该花器官细胞身份调控网络大约代表整个网络中的一个子网;所以虽然对于全局花发育基因调控网络而言,进化速率受到了基因中心性的影响,但同时又受到了局部功能约束的影响。如Szedlak等[21]研究发现,人类基因调控网络中,基因的进化特性与节点的中心性度量相关,同时在基因聚类簇内部的进化速率却相对均一[21]。
3 结论与讨论
该研究探讨了在花发育基因调控网络中每个基因的网络拓扑中心性包括连接度、中间性和接近度对其编码区序列进化速率的影响。结果发现,花发育基因调控网络总体上在纯化选择下进化,但随着网络中心性的增加,基因的序列将更加趋向于保守。这种趋势对于连接度、中间性和接近度而言方向都是相同的,只是在程度和显著性上略有差别。该研究为在网络背景下理解花发育相关基因的分子进化提供了新的数据。由于目前关于拟南芥花发育基因调控网络数据的数量和质量仍在不断发展中[1],因此该研究所得出的相关性结论也受限于所选用的调控网络数据来源。另外,在实际的生物系统中,除网络拓扑中心性外,有可能还有其他生物学参数也影响了基因序列的进化,如表达水平和功能类别。因此,在今后的相关性研究中,随着可用数据信息增加,可以研究更多的基因和生物学参数,以便进一步地了解花发育调控网络的保守性和可进化性。
參考文献
[1]LEMOS B,BETTENCOURT B R,MEIKLEJOHN C D,et al.Evolution of proteins and gene expression levels are coupled in Drosophila and are independently associated with mRNA abundance,protein length,and number of protein-protein interactions [J].Molecular biology and evolution,2005,22(5):1345-1354.
[2]FRASER H B,HIRSH A E,STEINMETZ L M,et al.Evolutionary rate in the protein interaction network [J].Science,2002,296(5568):750-752.
[3]HAHN M W,KERN A D.Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks [J].Molecular biology and evolution,2005,22(4):803-806.
[4]MASALIA R R,BEWICK A J,BURKE J M.Connectivity in gene coexpression networks negatively correlates with rates of molecular evolution in flowering plants [J].PLoS One,2017,12(7):1-10.
[5]JORDAN I K,MARIO-RAMREZ L,WOLF Y I,et al.Conservation and coevolution in the scale-free human gene coexpression network [J].Molecular biology and evolution,2004,21(11):2058-2070.
[6]JOVELIN R,PHILLIPS P C.Evolutionary rates and centrality in the yeast gene regulatory network [J].Genome biology,2009,10(4):1-10.
[7]PAJORO A,BIEWERS S,DOUGALI E,et al.The(r)evolution of gene regulatory networks controlling Arabidopsis plant reproduction:A two-decade history [J].Journal of experimental botany,2014,65(17):4731-4745.
[8]LIU Y,GUO C C,XU G X,et al.Evolutionary pattern of the regulatory network for flower development:Insights gained from a comparison of two Arabidopsis species [J].Journal of systematics and evolution,2011,49(6):528-538.
[9]DAVILA-VELDERRAIN J,SERVIN-MARQUEZ A,ALVAREZ-BUYLLA E R.Molecular evolution constraints in the floral organ specification gene regulatory network module across 18 angiosperm genomes[J].Molecular biology and evolution,2014,31(3):560-573.
[10]BERARDINI T Z,REISER L,LI D,et al.The Arabidopsis information resource:Making and mining the “gold standard” annotated reference plant genome [J].Genesis,2015,53(8):474-485.
[11]CAMACHO C,COULOURIS G,AVAGYAN V,et al.BLAST+:Architecture and applications [J].BMC Bioinformatics,2009,10(1):1-9.
[12]HAGBERG A A,SCHULT D A,SWART P J.Exploring network structure,dynamics,and function using networkx [C]//VAROQUAUX G,VAUGHT T,MILLMAN J,et al.Proceedings of the 7th Python in Science Conference.Pasadena,CA USA:[s.n.],2008.
[13]KATOH K,STANDLEY D M.MAFFT multiple sequence alignment software version 7:Improvements in performance and usability [J].Molecular biology and evolution,2013,30(4):772-780.
[14]SUYAMA M,TORRENTS D,BORK P.PAL2NAL:Robust conversion of protein sequence alignments into the corresponding codon alignments [J].Nucleic acids research,2006,34:W609-W612.
[15]CASTRESANA J.Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis [J].Molecular biology and evolution,2000,17(4):540-552.
[16]GUINDON S,DUFAYARD J F,LEFORT V,et al.New algorithms and methods to estimate maximum-likelihood phylogenies:Assessing the performance of PhyML 3.0 [J].Systematic biology,2010,59(3):307-321.
[17]LEFORT V,LONGUEVILLE J E,GASCUEL O.SMS:Smart model selection in PhyML [J].Molecular biology and evolution,2017,34(9):2422-2424.
[18]YANG Z.PAML 4:Phylogenetic analysis by maximum likelihood [J].Molecular biology and evolution,2007,24(8):1586-1591.
[19]ALVAREZ-PONCE D,FARES M A.Evolutionary rate and duplicability in the Arabidopsis thaliana protein-protein interaction network [J].Genome biology and evolution,2012,4(12):1263-1274.
[20]HE X,ZHANG J.Toward a molecular understanding of pleiotropy [J].Genetics,2006,173(4):1885-1891.
[21]SZEDLAK A,SMITH N,LIU L,et al.Evolutionary and topological properties of genes and community structures in human gene regulatory networks [J].PLoS Computational Biology,2016,12(6):1-16.
