
一步法制备富氧木质素活性炭及其亚甲基蓝吸附性能
2021-06-03 07:47王晶韩巧宁雷以廷唐曼陈丽红车俊达刘祖广
化工学报 2021年5期
王晶,韩巧宁,雷以廷,唐曼,陈丽红,车俊达,刘祖广
(1广西民族大学化学化工学院,广西南宁530006;2广西林产化学与工程重点实验室,广西林产化学与工程协同创新中心,广西南宁530006)
引 言
活性炭是一种最常用的吸附剂、催化剂及催化剂载体。它发达的孔隙结构、巨大的比表面积和丰富的表面功能基团,使其具有优异的吸附性能,广泛应用于环保、生物医药、化工、冶金、能源储存等各个领域[1-7]。活性炭的比表面积、孔隙结构和表面功能基团对其吸附性能有重要影响。比表面积和孔隙结构的调控通常是通过控制炭活化过程来实现的,而表面功能基团则主要是通过对活性炭的进一步改性而获得的。常用的活性炭表面改性方法有物理、化学改性、热处理和浸渍等[8-11],其中化学改性是调控活性炭表面官能团和化学特性的重要方法,如通过硝酸、过硫酸盐、过氧化氢、臭氧在一定条件下对活性炭进行氧化处理可以增加活性炭表面的羧基、羰基、酚羟基、内酯基等酸性含氧官能团,增强活性炭的表面亲水性及其对金属离子和极性物质的吸附能力,这种氧化改性方法虽然能够有效增加活性炭的表面含氧官能团,但通常会破坏活性炭原有的孔隙结构,降低活性炭的比表面积和总孔体积[10,12-16],从而对其性能造成一定的影响。此外,活性炭的氧化改性是在活性炭制造完成后的表面再改性,生产工艺冗长,原料损耗和生产成本相对增加。因此,对表面富氧活性炭的制备技术仍有待于进一步研究。
本实验将活性炭的制备与氧化改性合二为一,采用磷酸、硫酸组成的混合酸为活化剂,一步制备具有高比表面积和丰富含氧官能团的木质素活性炭。混合酸既起到活化的作用,又达到氧化改性的目的,得到比单一磷酸活化比表面积更大、表面氧含量更高、孔径分布更集中的微介孔活性炭,简化了表面富氧活性炭的制备工艺,克服了氧化改性活性炭的不足,具有良好的实际应用前景。
1 实验材料和方法
1.1 材料
木质素(纯度94.82%;灰分2.33%;苯乙醇抽提物2.85%),从南宁凤凰造纸厂提供的造纸黑液中提取;亚甲基蓝,分析纯(98%),上海麦克林生化科技有限公司;其他试剂均为市售分析纯试剂。
1.2 木质素活性炭的制备
磷酸活化木质素活性炭的制备:将木质素研细后加入浸渍比(磷酸与木质素的浸渍质量比)分别为1.0、2.0、3.0或4.0的40%磷酸溶液中浸渍12 h,然后将这些混合物在120℃下干燥24 h,并在氮气保护下在管式电阻炉中加热至400℃活化3 h,然后冷却到室温,将活化料用去离子水浸泡并反复洗涤直至滤液pH与洗涤水相同时为止。将得到的黑色固体于120℃下干燥6 h即得磷酸活化活性炭,表示为LAC400-Px(其中“400”表示炭活化温度为400℃,“x”是H3PO4的浸渍比)。
磷酸和硫酸混合酸活化木质素活性炭的制备:将上述制备方法中的40%磷酸溶液改为一定量40%磷酸和50%硫酸组成的混合酸溶液,其余制备过程同上。所获得的活性炭表示为LAC400-Py-Sz(其中“y”为H3PO4与木质素的质量比,“z”为H2SO4与木质素的质量比)。
1.3 木质素活性炭的性能表征
1.3.1 活性炭活化酸利用率的测定 采用酸碱滴定法[17]测定木质素活化反应后残余强酸,用参与活化消耗的总酸与初始总酸的比值来表示酸利用率。因硫酸盐木质素本身含有少量的硫,其在活化过程中变化复杂,难以确定回收酸中硫的来源,因此本实验未将硫酸和磷酸分开测定,仅测定总酸的利用率。将上述1.2节制备的混合酸活化木质素活性炭用去离子水反复洗涤至滤液呈中性,收集多次洗涤后的滤液于容量瓶并定容至500 ml,取25.00 ml于锥形瓶中,以百里香酚酞为指示剂,用氢氧化钠标准液进行滴定至指示剂由无色变为浅蓝色即为终点(磷酸滴定到磷酸氢二钠状态),计算酸利用率。
1.3.2 活性炭表面酸性官能团含量测定 采用Boehm滴定法测定木质素活性炭表面酸性官能团的含量[18-19]。精确称取三份0.2500 g待测样品分别放入三个50 ml烧杯中,然后分别加入0.0500 mol·L-1氢氧化钠标准溶液、0.5000 mol·L-1氯化钠溶液和0.0500 mol·L-1碳酸氢钠标准溶液各25.00 ml,密封,在室温下超声1 h后过滤、洗涤,将滤液完全转移至锥形瓶中,加入1~2滴1%酚酞乙醇溶液指示剂,用0.0500 mol·L-1盐酸标准溶液滴定至红色刚刚消失,或者用0.0500 mol·L-1氢氧化钠标准液滴定至刚刚出现微红色且30 s内不褪色即为滴定终点,计算酸值。重复测定三次,以平均值作为测定结果。酸性基团含量的测定是基于以下假定:氢氧化钠中和强酸、羧酸、内酯基团和酚羟基,氯化钠只交换强酸中质子,碳酸氢钠中和强酸和羧酸。
1.3.3 红外光谱测试方法 在玛瑙研钵中将少量待测样品和适量KBr粉末混合均匀磨细后压片,将压片后的样品置于红外光谱仪(Bruker Vector 33)上进行测试,在4500~400 cm-1范围内进行扫描。
1.3.4 扫描电镜测试方法 将待测样品烘干,取少量并将其黏附在导电胶上,固定在样品台上,抽真空后在场发射扫描电子显微镜(SUPRA 55)上观察表面结构并拍照。
1.3.5 Zeta电位测试 分别用1.0 mol·L-1HCl和1.0 mol·L-1NaOH溶液调节蒸馏水pH分别为1.0、2.0、3.0、4.0、5.0、6.0、7.0、8.0、9.0、10.0、11.0和12.0,然后分别加入0.0100 g活性炭,室温超声1 h,再用酸或碱调节混合液的pH至原来水溶液的pH并定容至50 ml,混匀,用Zeta电位仪(Malvern Zetasizer Nano)测定Zeta电位。
1.3.6 接触角测试方法 以水为介质用接触角测定仪(JY-82B)进行接触角测试。待测样品磨细后压片,将水滴滴于样品表面,通过显微镜头与相机获得液滴的外形图像,再运用数字图像处理和Young-Laplace方程计算图像中的液滴接触角。
1.3.7 氮气吸附脱附测试 氮气吸附脱附实验在比表面积和孔径分析仪(Micromeritics ASAP 2020)上进行。测试温度在77 K下进行,将材料置于氮气气氛下进行吸附测试。实验前,样品在120℃、氮气气氛下脱气12 h。
1.4 木质素活性炭吸附亚甲基蓝
移取50.00 ml浓度为200 mg·L-1的亚甲基蓝溶液到100 ml的锥形瓶中,加入一定量的木质素活性炭,密封,在150 r·min-1的水浴恒温摇床上于一定温度下振荡吸附,每间隔一定时间以0.22μm的膜滤器过滤取样,并用UV-Vis分光光度计(Agilent Cary 60)在664 nm处测定剩余亚甲基蓝的吸光度,根据亚甲基蓝溶液的工作曲线得到其浓度,并计算吸附量。为确保吸附达到平衡,平衡吸附量的振荡吸附时间为24 h。
2 实验结果与讨论
2.1 木质素活性炭的表征
2.1.1 扫描电镜分析 木质素炭化样、磷酸活化活性炭及混合酸活化活性炭的扫描电镜如图1所示。由图可见,未添加磷酸活化的木质素炭化样LAC400-P0表面平整光滑,未见明显的孔隙结构;而添加磷酸样品LAC400-P2.0、LAC400-P3.0和LAC400-P4.0均呈现多孔的珊瑚状结构。与LAC400-P3.0相比,LAC400-P4.0活性炭的颗粒变大。磷酸和硫酸混合酸活化活性炭与磷酸活化木质素活性炭形貌明显不同,前者并不呈珊瑚状结构,而是表面凹凸不平,存在许多微小的孔隙结构。以上结果表明,直接炭化木质素未能得到多孔结构;而采用磷酸浸渍可有效地起到活化作用,得到多孔的珊瑚状结构活性炭。但当磷酸浸渍比达到4.0时,所得活性炭表面颗粒变大,可能不利于比表面积的提高。混合酸活化活性炭,由于硫酸的存在可能影响了活化过程中磷酸及其衍生物形成大的聚集体,结果得到分布更趋集中的孔隙结构,从而有利于得到性能更好的活性炭。
2.1.2 氮气吸附脱附分析 磷酸活化木质素活性炭及混合酸活化木质素活性炭的氮气吸附脱附曲线及孔径分布图如图2所示,其氮气吸附脱附等温线[图2(a)]均表现为Ⅳ型吸附,表明这两种活性炭均为介孔碳材料。曲线伴有的回滞环为H3型多孔吸附行为,且在相对压力P/P0接近1时,没有表现出吸附限制性,说明样品具有堆砌的狭缝状孔隙[20]。磷酸活化活性炭的氮气吸附量随着磷酸浸渍比的增加先增加后下降,混合酸活化样品具有更大的吸附量,表明其具有更大的比表面积。活性炭孔径分布[图2(b)]表明,不同浸渍比磷酸活化活性炭具有相似的孔径分布趋势,从微孔到大孔均有一定分布,孔径分布宽;而混合酸活化活性炭,孔径分布更趋集中,绝大多数孔径在30 nm以下,其中以3 nm左右的孔隙分布最多,这与扫描电镜观察的结果相一致。

图1 木质素炭化样LAC400-P0(a),磷酸活化木质素活性炭LAC400-P2.0(b)、LAC400-P3.0(c)、LAC400-P4.0(d)和混合酸活化木质素活性炭LAC400-P3.0-S0.5(e)的扫描电镜Fig.1 SEMimagesof LAC400-P0(a),LAC400-P2.0(b),LAC400-P3.0(c),LAC400-P4.0(d)and LAC400-P3.0-S0.5(e)
从表1可见,经过磷酸活化的木质素活性炭具有较大的比表面积和丰富的孔隙结构。当磷酸与木质素的浸渍比为3.0时,比表面积844 m2·g-1、总孔体积0.87 cm3·g-1和平均孔径6.57 nm均达到最大值,此时介孔率也达到最大值86.2%;而混合酸活化活性炭的比表面积进一步提高到1080 m2·g-1,总孔体积达到1.10 cm3·g-1,分别比磷酸活化木质素活性炭增加了27.9%和26.4%,介孔体积也增大了29.3%。这可能是在炭活化时,由于硫酸和磷酸的协同催化作用,进一步促进了木质素与磷酸之间的酯化交联反应,从而有效地防止了因炭化收缩而引起的孔隙塌陷[21-22]。此外,炭活化后水洗除去磷酸及其衍生物产生的大量孔隙结构[23],是活性炭孔隙的主要来源。如果磷酸用量过多,磷酸分子可能会形成一些尺寸更大、分布更宽的聚集体,导致以此为骨架形成的活性炭的颗粒变大、孔径分布变宽,其结果是比表面积和总孔体积反而下降。而混合酸活化活性炭,由于硫酸的存在,可能在一定程度上减小了磷酸的聚集尺寸,使得所形成的孔隙尺寸更加均匀而集中,主要产生介孔和微孔,因而比表面积、总孔体积和介孔率进一步增加。以上表征表明,采用混合酸活化活性炭有利于获得孔隙分布更集中、比表面积更大的微介孔活性炭。

图2 磷酸、混合酸活化木质素活性炭的氮气吸附脱附等温线(a)和孔径分布图(b)Fig.2 N2 adsorption-desorption isotherms of the carbons activated by phosphoric acid and mixed acid,respectively(a)and their pore size distribution curves(b)
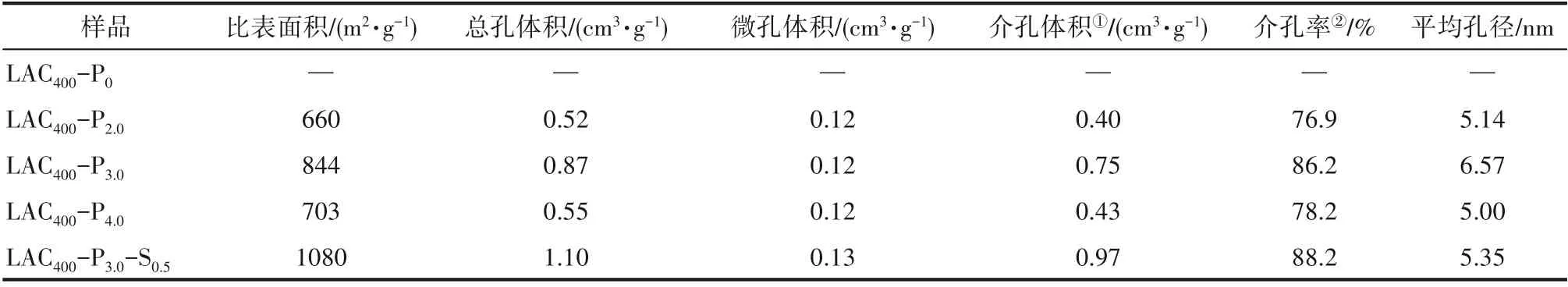
表1 木质素炭化样及木质素活性炭的结构性质Table 1 Surface and textural characteristics of kraft lignin activated carbon
2.1.3 红外光谱分析 木质素、磷酸活化木质素活性炭、混合酸活化木质素活性炭的红外光谱图如图3所示。图中,3420 cm-1和1705 cm-1附近的吸收峰分别属于—OH和C O的伸缩振动,表明活性炭中存在羟基和羧基[24]。1587 cm-1的吸收峰归属于芳香环或C C的伸缩振动[25]。1220~1180 cm-1区域的吸收峰对应于与氢结合的P O键、P-O-C(芳环)中O—C和P OOH化学键的伸缩振动[26]。1090 cm-1处的吸收峰是由于磷酸酯中P+-O-离子伸缩振动和聚磷酸盐中P-O-P的不对称伸缩振动产生的[27]。与LAC400-P3.0相比,混合酸活化活性炭LAC400-P3.0-S0.5在1210 cm-1和1090 cm-1处吸收峰增强,表明后者存在更多的磷酸酯[28];在波数为1587 cm-1的C C吸收峰明显增强,可能是硫酸的加入促进木质素的脱水炭化和芳环化的缘故[29];并且在3420 cm-1和1705 cm-1处—OH和C O的吸收峰强度明显增强,说明LAC400-P3.0-S0.5中含氧官能团含量明显增加,这与后面元素分析及酸碱滴定得到的结果相一致。
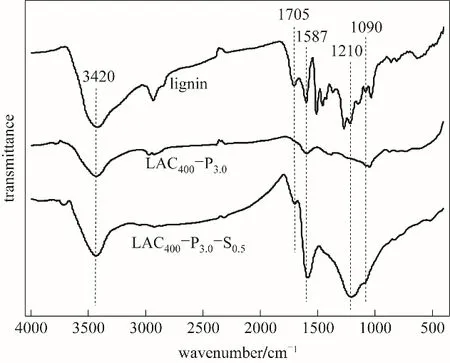
图3 木质素、磷酸活化木质素活性炭和混合酸活化木质素活性炭的红外光谱图Fig.3 FTIRspectra of lignin,LAC400-P3.0 and LAC400-P3.0-S0.5
2.1.4 表面化学性质分析 采用Boehm滴定法测定磷酸和混合酸活化木质素活性炭结果如表2所示。磷酸活化活性炭的总酸含量为2.46~2.68 mmol·g-1,而混合酸活化的活性炭总酸含量明显提高,达到了3.35 mmol·g-1,这是由于硫酸的氧化作用,使LAC400-P3.0-S0.5酚羟基(包括内酯基团)含量显著增加,且提升了活性炭的亲水性能(LAC400-P3.0-S0.5接触角仅为4.16°,而LAC400-P3.0的接触角则为14.3°)。此外,磷酸活化的活性炭由于残留了一部分结合磷酸,因而具有一定的强酸酸值,这与红外光谱的结果相一致。
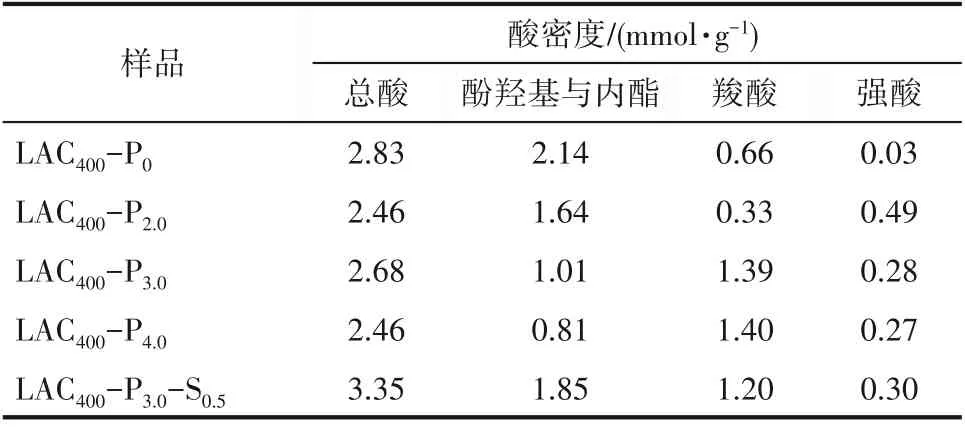
表2 木质素活性炭的酸密度Table 2 The acid density and adsorption capacity of LAC400-P x and LAC400-P3.0-S0.5
采用元素分析测定木质素及其活性炭的化学组成如表3所示。木质素在400℃炭活化后,碳含量增加,氢、氧、硫含量降低。由于加入磷酸催化了木质素的脱水反应[30],改变了热解历程,故所得活性炭的产率提高,而氧、氢含量继续降低。混合酸活化活性炭,由于硫酸的强氧化作用,其氧含量(24.45%)比磷酸活化活性炭氧含量(18.92%)提高了29.2%,表明其酸性含氧官能团明显增加,这与红外光谱和酸值滴定的结果相一致。同时,硫含量降低,可能是由于硫酸与木质素的结合硫发生了氧化还原反应生成二氧化硫而挥发造成的。
氧化活性炭由于其表面丰富的含氧官能团,使其具有良好的亲水性、络合能力和分子间作用力,对金属离子、极性物质和阳离子染料具有良好的吸附能力,是一类重要的改性活性炭。但氧化活性炭的制备通常都是采用碳前体先炭化、活化制备活性炭,再通过氧化改性制备氧化活性炭的方法。这种两步制备方法,虽然可以提高活性炭表面酸性含氧官能团,但对活性炭的孔隙结构造成较大破坏,导致其比表面积、总孔体积、微孔体积等均有不同程度的降低(表4),相对降低了活性炭的吸附能力。并且在氧化改性过程中难以有效控制活性炭的孔隙结构。Huang等[10]采用3 mol·L-1硝酸在100℃氧化活性炭,尽管活性炭的酸值有显著提升,但比表面积却由原来的2623 m2·g-1锐减到468 m2·g-1,微孔体积也从0.72 cm3·g-1降低到0.28 cm3·g-1;Shim等[12]采用1 mol·L-1稀硝酸氧化沥青基活性炭纤维2 h后,发现该活性炭纤维比表面积由1462 m2·g-1降低到976 m2·g-1,总孔体积由0.70 cm3·g-1降到0.48 cm3·g-1;Forouzesh等[13]采用浓硝酸氧化活性炭后,同样发现氧化活性炭的比表面积、总孔体积、微孔体积都有明显下降,平均孔径增加;Kampouraki等[14]分别采用硝酸、硫酸氧化活性炭,发现氧化活性炭的比表面积、总孔体积、微孔体积和介孔体积均下降。本文以磷酸和硫酸混合酸为活化剂,采用一步法制备了含氧官能团丰富的活性炭,与单独磷酸活化的活性炭相比,不但没有降低比表面积,其比表面积、总孔体积、介孔体积和氧含量反而分别增加了27.9%、26.4%、29.3%和29.2%,并且孔径分布更趋集中,介孔率进一步提高,有利于提升活性炭的液相吸附能力。这种一步制备富氧活性炭的方法,不仅易于调节孔隙结构,提升活性炭性能,还简化了制备工艺、节约了生产成本。尽管本实验采用的磷酸和硫酸用量较大,但由于活化后回收的酸可重复利用,因此,真正用于活化消耗的酸量并不大。经过实验测定,回收酸量占总酸的53.72%,混合酸的利用率为46.28%,相当于1.00 g木质素仅消耗1.62 g混合酸。2.1.5 混合酸活化木质素的可能机理 尽管混合酸活化木质素活性炭的机理复杂,但总结以上研究结果,并结合磷酸活化机理[31],认为混合酸的活化机理可能有以下几个方面:首先,由于混合酸中硫酸的催化作用,进一步促进了磷酸对木质素的脱水炭化、芳香缩合和酯化交联作用,从而防止了木质素因炭化收缩而引起的孔隙塌陷。其次,混合酸中由于硫酸与碳发生氧化还原反应,一方面侵蚀炭体起到了造孔和扩孔作用,另一方面使活性炭的表面含氧官能团增加。另外,反应所产生的二氧化碳和二氧化硫也会有致孔作用,因而混合酸活化活性炭的比表面积、总孔体积、氧含量与单独磷酸活化活性炭相比有显著提高。第三,混合酸中硫酸可能在一定程度上减少了磷酸的聚集尺寸和颗径分布,因而使得以其为骨架沉积生成的活性炭主要产生微孔和介孔,并且孔径分布更趋集中,也会使比表面积和介孔率比单独磷酸活化活性炭进一步提升。

表3 木质素活性炭化学组成及炭产率Table 3 Chemical composition measured by elemental analysis and carbon yield of the activated carbon
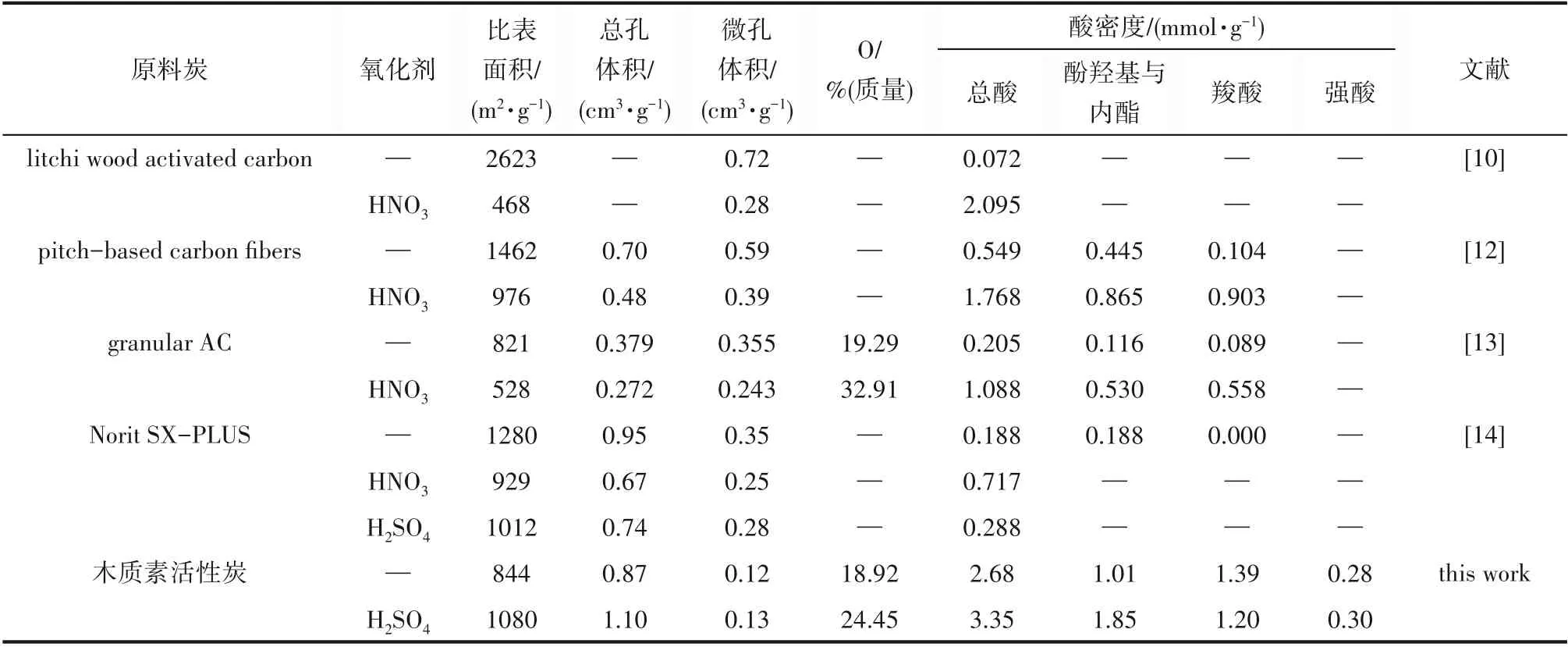
表4 不同活性炭氧化前、后的比表面积、孔结构特征、酸密度等变化情况Table 4 Comparison of the specific surface area,textural properties and acid density of activated carbons before and after oxidation
当然,由于混合酸与木质素的反应十分复杂,相关活化机理还有待于进一步深入研究。总之,混合酸既充当了活化剂的作用,又起到了氧化剂的作用,使其活化的活性炭的性能显著高于单独磷酸活化的活性炭。
2.2 对亚甲基蓝的吸附性能
2.2.1 活化剂的影响 表5为不同木质素炭材料对亚甲基蓝的吸附能力。由表可见,未经过活化的木质素炭材料由于比表面积基本可以忽略不计,因而对亚甲基蓝的吸附量少。经过磷酸活化的木质素活性炭,由于比表面积大,并且在活化过程中引入了部分磷酸功能基团,故吸附能力急剧增加。而经过混合酸活化的木质素活性炭,由于比表面积与表面含氧功能基团的进一步增加,其亚甲基蓝的吸附能力进一步提高到1118.90 mg·g-1,比磷酸活化样提升了20.3%。

表5 木质素炭材料对亚甲基蓝的吸附能力Table 5 The adsorption capacity of carbon materials derived from kraft lignin to MB
2.2.2 投加量的影响 混合酸活化木质素活性炭投加量对初始浓度为200 mg·L-1亚甲基蓝吸附性能的影响如图4所示。随着活性炭投加量的增加,亚甲基蓝的去除率不断增加。当投加量达到0.0150 g·L-1后,继续增加活性炭投加量去除率仅略有增加,说明此时基本达到吸附平衡。此外,随着投加量增加,活性炭的平衡吸附量(qe)不断地减小,当投加量在0.01 g·L-1之前下降比较缓慢,之后下降幅度较大,且此时去除率的增加趋缓,因此选择0.01 g·L-1为合适的投加量,不仅避免了资源浪费,又可以得到较好的吸附效果。此时,亚甲基蓝的去除率为81.76%,活性炭的吸附量达到937.60 mg·g-1。

图4 LAC400-P3.0-S0.5投加量的影响(吸附条件:T=298 K;C0=200 mg·L-1;pH=7)Fig.4 Effect of dosage adsorbent(adsorption conditions:T=298 K;C0=200 mg·L-1;pH=7)
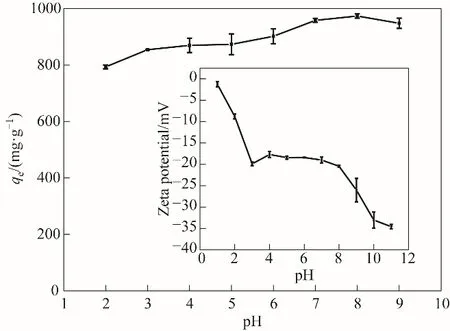
图5 溶液pH对木质素活性炭吸附亚甲基蓝和Zeta电位(插图)的影响(吸附条件:T=298 K)Fig.5 Effect of solution pH on the MBadsorption by LAC400-P3.0-S0.5 and Zeta potential as a function of pH(inset)(adsorption conditions:T=298 K)
2.2.3 溶液pH的影响 溶液pH对吸附性能的影响如图5所示。亚甲基蓝在活性炭LAC400-P3.0-S0.5上的吸附量随着溶液pH的增加而增加。Zeta电位测量表明,活性炭LAC400-P3.0-S0.5在整个测试pH范围内表面均带负电,并且随着溶液pH的变化而变化,这可能是活性炭表面的不同酸性官能团的电离引起的。pH在1~3范围内,随着pH的升高,活性炭的负电荷急剧增强,可能是由于表面强酸和羧基的不断电离造成的;pH在3~8范围内,由于强酸和羧基的电离基本趋于完全,故负电荷基本趋于稳定;pH在8~10范围内,由于酚羟基的不断离解,所以随着pH的升高表面负电荷又急剧增加;pH在10~11范围内,由于酚羟基的离解接近完全,故表面负电荷仅略有增加。由于该活性炭在整个测试pH范围内均带负电荷,故对带正电性的亚甲基蓝染料有良好的吸附性能,随pH升高,对亚甲基蓝吸附能力增强。但是,当pH增大到9时,对亚甲基蓝的吸附量反而略有下降,可能是因为溶液中OH-浓度增加,与活性炭表面的负电荷的竞争能力增加,导致更多的亚甲基蓝以分子形式存在于溶液中,从而降低了活性炭表面对亚甲基蓝的吸附能力[32]。
2.3 Langmuir吸附等温过程
采用Langmuir等温线模型研究了混合酸活化木质素活性炭对亚甲基蓝的吸附特性。Langmuir等温吸附方程如下:
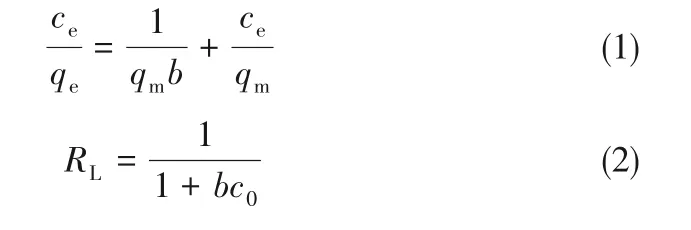
式中,qe为吸附剂的平衡吸附量,mg·g-1;ce为溶液中吸附质的平衡浓度,mg·L-1;qm为吸附剂的最大吸附容量,mg·g-1;b为Langmuir吸附常数,L·mg-1;RL为无量纲常数分离因子。
LAC400-P3.0-S0.5对亚甲基蓝的吸附等温线如图6所示。其对亚甲基蓝的吸附量随着亚甲基蓝初始浓度的增加而增加[图6(a)]。该吸附等温线的线性拟合[图6(b)]表明,LAC400-P3.0-S0.5对亚甲基蓝的吸附符合Langmuir吸附模型(R2=0.995,b=0.64 L·mg-1,qm=1022.46 mg·g-1),这表明对亚甲基蓝的吸附为均匀的单分子层吸附[33]。而且,0<RL(0.0044~0.01)<1,说明该吸附为优惠吸附。
2.4 吸附动力学
为了测定LAC400-P3.0-S0.5对亚甲基蓝吸附动力学,测定了混合酸活化活性炭吸附亚甲基蓝的动力学曲线,并采用伪一级、伪二级动力学方程和粒子内扩散模型对吸附过程进行了研究。伪一级和伪二级动力学分别表示为式(3)和式(4)。
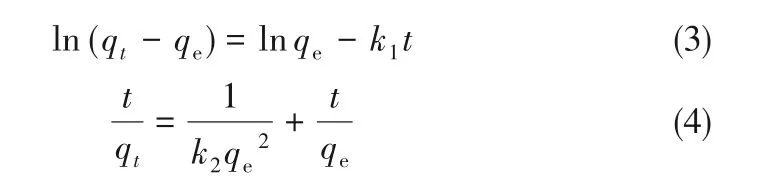
式中,k1为伪一级动力学速率常数,min-1;k2为伪二级动力学速率常数,g·mg-1·min-1;t为接触时间,min;qt为t时刻的吸附量,mg·g-1。颗粒内扩散模型如式(5)所示。

式中,kp为颗粒内扩散速率常数,mg·g-1·min-1/2;C为与边界层厚度有关的常数,mg·g-1。
LAC400-P3.0-S0.5的吸附动力学曲线如图7所示。图7(a)显示了接触时间对LAC400-P3.0-S0.5吸附亚甲基蓝(初始浓度为200 mg·L-1)的影响。由图可见,初期吸附过程十分迅速,在15 min内亚甲基蓝被从溶液中除去了约50%,并在4 h后接近吸附平衡。亚甲基蓝在LAC400-P3.0-S0.5上的快速吸附可以归因于其较大的表面积、丰富的微介孔结构和强烈的表面作用力。LAC400-P3.0-S0.5吸附亚甲基蓝的动力学参数见表6。图7(b)、(c)分别为吸附过程的伪一级动力学、伪二级动力学拟合。由于伪二级动力学模型的相关系数(R2=0.99)高于伪一级动力学模型的相关系数(R2=0.97),说明吸附过程更符合伪二级动力学方程,表明吸附过程涉及化学效应[34]。并且,根据伪二级动力学理论计算的平衡吸附容量值(qe,cal=1024.49 mg·g-1)与实际实验所测定的吸附容量值(qe,exp=1118.90 mg·g-1)具有极好的一致性。

图6 LAC400-P3.0-S0.5对亚甲基蓝的吸附等温线(吸附条件:T=298 K,pH=8)(a)初始浓度的影响;(b)Langmuir线性拟合Fig.6 Adsorption isotherm of LAC400-P3.0-S0.5 on MB(adsorption conditions:T=298 K;pH=8)(a)effect of dyeinitial concentration and temperature;(b)Langmuir linear fit
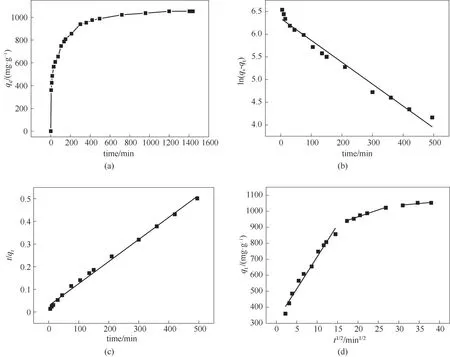
图7 木质素活性炭对亚甲基蓝吸附的动力学曲线(吸附条件:T=298K;pH=8)(a)接触时间的影响;(b)伪一级动力学拟合;(c)伪二级动力学拟合;(d)颗粒内扩散模型Fig.7 Kinetics curves for the MBadsorption by LAC400-P3.0-S0.5(adsorption conditions:T=298 K;pH=8)(a)effect of thecontact time;(b)fittingof pseudo-first-order kinetics;(c)fittingof pseudo-second-order kinetics;(d)intra-particlediffusion model
颗粒内扩散模型的qt与t1/2之间的关系表明,LAC400-P3.0-S0.5吸附亚甲基蓝的过程涉及三个线性阶段[图7(d)]。第一阶段代表膜/表面扩散,其中亚甲基蓝分子扩散通过液膜并迅速吸附到LAC400-P3.0-S0.5的外表面。此阶段具有最高的颗粒内扩散速率常数(kp)和相对较低的C值(表6),表明LAC400-P3.0-S0.5具有足够的表面积和活性位点[35]。在粒子内扩散的第二阶段,亚甲基蓝分子扩散进入LAC400-P3.0-S0.5的孔隙中[36]。第三阶段代表亚甲基蓝分子在LAC400-P3.0-S0.5内表面上达到吸附平衡,具有最低的kp,这是由于此时溶液中残留的亚甲基蓝浓度较低,并且活性炭表面几乎没有剩余的游离活性位点,导致亚甲基蓝的扩散速率降低。此外,方程未通过原点,表明粒子内扩散不是唯一的速率控制步骤[37]。

表6 木质素活性炭LAC400-P3.0-S0.5对亚甲基蓝吸附的伪一级、伪二级动力学及颗粒内扩散参数Table 6 Pseudo-first-order,pseudo-second-order kinetics and intra-particle diffusion parameters for the adsorption of MB on LAC400-P3.0-S0.5
3结 论
(1)分别以磷酸、磷酸和硫酸混合酸为活化剂制备了两种木质素活性炭,在磷酸浸渍比3.0,炭活化温度为400℃和炭活化时间为3 h的条件下,制备得到的磷酸活化木质素活性炭具有最佳的综合性能。在此基础上,通过添加质量为0.5倍木质素的硫酸到磷酸中组成混合酸活化剂,制备出比表面积达1080 m2·g-1、总孔体积为1.10 cm3·g-1、介孔体积为0.97 cm3·g-1、总酸密度为3.35 mmol·g-1的活性炭。它比磷酸活化的木质素活性炭比表面积增加了27.9%,总孔体积增加了26.4%,介孔体积增加了29.3%,孔径分布在40 nm以下,并且氧含量增加了29.2%,为一种含氧官能基团丰富的微介孔活性炭。
(2)混合酸活化的木质素活性炭对模拟废水中的亚甲基蓝在pH为2~9的范围内均具有优异的吸附性能,在pH=7时,最大饱和吸附量达到1118.90 mg·g-1,比单独磷酸活化木质素活性炭吸附量增加了20.3%。对初始浓度为200 mg·L-1的亚甲基蓝溶液,当活性炭的投加量为0.0100 g·L-1时,亚甲基蓝的吸附去除率为81.76%,吸附量达到937.60 mg·g-1。吸附速率快,吸附过程遵循Langmuir等温线和伪二级动力学方程,颗粒内扩散不是唯一的决速步骤。
猜你喜欢
造纸信息(2022年8期)2022-11-10
轮胎工业(2021年4期)2021-12-25
新疆钢铁(2021年1期)2021-10-14
磷肥与复肥(2021年8期)2021-09-28
铁道建筑技术(2021年4期)2021-07-21
化工管理(2021年7期)2021-05-13
有色设备(2021年4期)2021-03-16
无机盐工业(2020年11期)2020-11-21
新能源进展(2020年1期)2020-03-09
中国造纸(2019年6期)2019-09-10
