
基于癌症基因组图谱计划多组学数据构建胶质母细胞瘤六基因预后模型
2021-07-28 06:41雷常贵贾学渊孙文靖
遗传 2021年7期
雷常贵,贾学渊,孙文靖
研究报告
基于癌症基因组图谱计划多组学数据构建胶质母细胞瘤六基因预后模型
雷常贵,贾学渊,孙文靖
哈尔滨医科大学医学遗传学研究室,中国遗传资源保护与疾病防控教育部重点实验室(哈尔滨医科大学),哈尔滨 150081
胶质母细胞瘤(glioblastoma, GBM)是最常见的原发性颅内肿瘤,恶性程度极高,患者预后极差。为了识别GBM预后生物标记物,建立预后模型,本研究通过分析癌症基因组图谱计划(The Cancer Genome Atlas, TCGA)数据库中GBM的表达谱数据,筛选出不同生存期GBM患者差异基因。利用GISTIC软件和Kaplan-Meier (KM)生存分析方法分析TCGA数据库中的GBM拷贝数变异数据,识别影响生存的扩增基因(survival-associated amplified gene, SAG)。取短生存期组上调基因和SAG两者的交集基因,进行单因素Cox回归和迭代Lasso回归筛选重要候选基因并建立预后模型;计算预后评分,根据预后评分中位数将患者分为高风险组和低风险组。用ROC曲线判断模型的优良,KM生存分析高低风险组预后差异,并用GEO、CGGA和Rembrandt数据库3个外部数据集进行验证。多因素Cox回归分析判断预后评分的预后独立性。结果显示,GBM不同生存期差异分析得到上调基因426个,下调基因65个。短生存期组上调基因与SAG交集得到47个基因。经过筛选,最终确定六基因(、、、、、)预后模型。TCGA实验组和3个外部验证组模型的ROC曲线下面积均大于0.6,甚至达到0.912。KM分析显示高低风险组的预后都存在差异(<0.05)。在多因素Cox回归分析中,六基因预后评分是GBM患者预后的独立影响因素(<0.05)。通过一系列分析,本研究确立了六基因(、、、、、)的GBM预后模型,模型具有很好的预测能力,可作为预测GBM患者的独立预后标志物。
胶质母细胞瘤;多组学数据;六基因组合;预后模型;癌症基因组图谱计划
胶质瘤(glioma)是最常见的原发性颅内肿瘤,占所有颅内肿瘤的80%[1]。世界卫生组织(World Health Organization, WHO) (2016年中枢神经系统肿瘤分类总结)将弥漫性胶质瘤分为WHO II级和III级星形细胞瘤、II级和III级少突胶质细胞瘤、IV级胶质母细胞瘤以及儿童相关的弥漫性胶质瘤[2]。不同级别的胶质瘤在肿瘤病理形态(如胶原纤维含量和形态多样性)、肿瘤发展和患者预后等方面差异很大[3]。其中胶质母细胞瘤(glioblastoma, GBM, WHO IV)最为常见,且患者预后最差,总生存期中位数仅为12~ 14个月;但是,有3%~5%的患者可以存活超过3年,被称为长期存活者[4]。与长期存活相关的临床和分子因素仍然匮乏,目前在弥漫性胶质瘤中仅有异柠檬酸脱氢酶(isocitrate dehydrogenases, IDH)突变状态、O6-甲基鸟嘌呤-DNA 甲基转移酶(O-6-methylguanine- DNA methyltransferase, MGMT)基因启动子甲基化和染色体1p和19q联合缺失被广泛认知[4]。GBM患者不同的生存期差异也反映出关键肿瘤相关基因表达水平或基因组改变存在不同[5]。
拷贝数变异(copy number variations, CNV)通常被定义为涉及大规模(>1 kb)基因组DNA变化的结构变异,CNV是基因表达的显著影响因素,可能影响多种致癌或抑癌关键通路的活性[6~9]。在胶质瘤中常见的拷贝数变异包括7号染色体片段的扩增,9号和10号染色体片段的缺失,以及在小范围内1号和19号染色体的缺失[10]。研究显示这些基因组结构变化主要与肿瘤相关基因的扩增,以及肿瘤抑制基因、和等的缺失或突变有关[3,11]。
各种变异对于GBM生存期的影响,目前GBM预后模型研究主要围绕转录组水平开展,如构建自噬相关的4个基因(和)的GBM预后模型[12],缺氧相关的5个基因(和)的GBM预后模型[13],免疫状态相关的15个基因的GBM预后模型[14]。另外通过筛选与GBM预后相关的4个miRNA (和)来建立预后模型[15]。在多组学层面,有研究通过对DNA甲基化和基因表达的综合分析识别关键标志物(和)来建立预后模型[16]。由于拷贝数变异的复杂性,目前对于GBM拷贝数变异相关的预后标志物的系统研究尚不多见,因此探讨不同生存期GBM之间的转录组水平差异,并结合GBM基因组层面的拷贝数变异数据,进行联合分析,将有助于更好地识别GBM的关键预后分子和潜在的治疗靶点。本研究对TCGA数据库中GBM患者的基因组数据和表达谱数据联合分析,建立了一个多基因预后模型,其在不同数据集中都能够很好地识别预后不良的高风险患者,为GBM患者的预后风险评估提供更好的依据。
1 材料与方法
1.1 数据收集
运用TCGAbiolinks包[17]下载癌症基因组图谱计划(The Cancer Genome Atlas, TCGA)数据库(https:// portal.gdc.cancer.gov/)中GBM患者的RNA-seq表达数据(GBM. RNAseq HtSeq Count, level 3),DNA拷贝数变异数据(GBM. SNP6.0 array),同时下载与患者相关临床数据。提取的临床数据信息包括总生存期(overall survival time, OS.time)、年龄、性别、基因突变状态等。我们用样本匹配RNA-seq数据,CNV数据和对应的临床数据提取了139个GBM患者数据用于分析[18]。
我们一共采用了3组数据作为验证模型。从美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)的高通量芯片表达谱数据库(Gene Expression Omnibus database, GEO, https://www.ncbi.nlm.nih.gov/geo/)中获取胶质瘤的GSE16011数据集,数据集中包含159个GBM样本,从中获取150个包含总生存期和生存状态信息的GBM样本作为验证组进行后续分析和模型验证。从中国脑胶质瘤图谱数据库(Chinese Glioma Genome Atlas, CGGA, http://www.cgga.org.cn/)下载了Part C的mRNAseq_325表达谱数据和临床数据,我们将GBM的88个样本数据作为验证组进行后续分析和模型验证。此外,我们还从美国脑肿瘤分子数据库(The Repository of Molecular Brain Neoplasia Data, Rembrandt, http://gliovis.bioinfo.cnio.es/)提取146个GBM样本的表达谱数据和临床数据作为验证组进行后续分析和模型验证。总生存期定义为从患者诊断日期开始至死亡日期截止。TCGA中GBM数据和GEO中GSE16011数据下载日期截止于2020年9月25日,CGGA数据和Rembrandt数据下载日期截止于2021年1月18日。
1.2 分析流程
本研究分析流程见图1所示。
1.3 不同生存期GBM患者表达谱差异分析
对于TCGA中139个GBM表达谱数据,将每个患者的总生存期小于1年(OS.time<1 year)作为短生存期患者,大于3年(OS.time>3 year)作为长生存期患者进行分组。运用Deseq2包[19]进行差异分析,筛选<0.05,并且绝对值Log2(fold change)>1作为短生存期组与长生存期组相比差异的显著基因。最终,通过火山图展示短生存期患者和长生存期患者的差异基因。
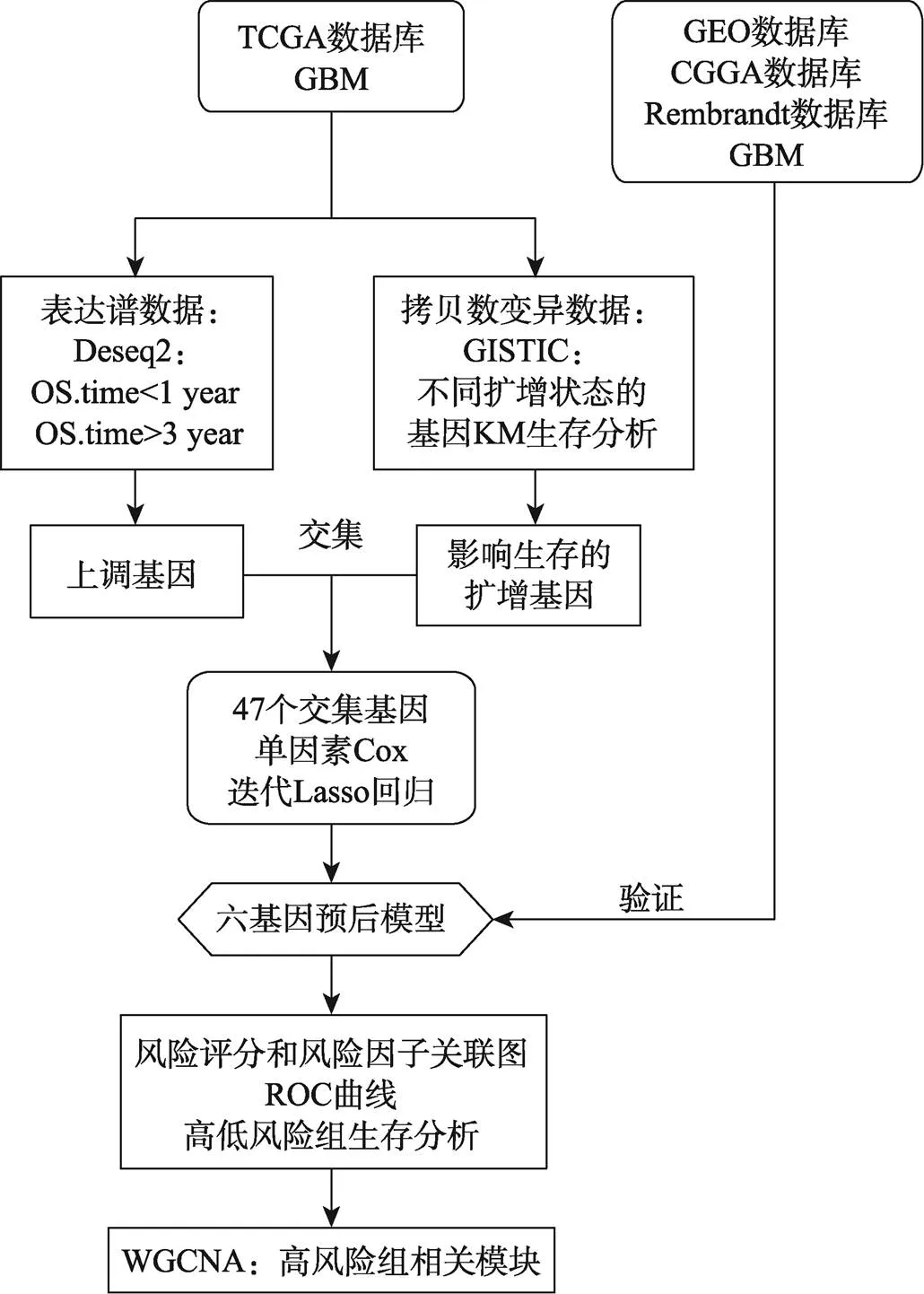
图1 分析流程图
1.4 GBM拷贝数变异数据分析
肿瘤中重要靶点的基因组识别(genomic identification of significant targets in cancer, GISTIC)是一种用于识别更有可能引发肿瘤发病机制的变异区域的算法[20]。利用GISTIC 2.0软件对139个GBM患者CNV数据进行分析,选择0.1作为拷贝数增加和缺失的阈值,-value=0.25作为显著性阈值,确定了GBM患者中所有基因的五种离散拷贝数状态(−2, −1, 0, 1, 2)。以基因的(1, 2)拷贝数作为基因扩增组,(−2, −1, 0)拷贝数作为基因非扩增组。
1.5 生存分析
Kaplan-Meier(KM)生存分析[21]用于分析基因与患者预后的关系。我们将GISTIC 2.0分析得到不同扩增状态的基因进行KM生存分析,筛选<0.05作为影响生存的扩增基因(survival-associated amplification gene, SAG)。
1.6 单因素Cox回归和迭代Lasso回归筛选候选基因
将差异分析中短生存期组上调基因和SAG取交集基因;通过survival包对交集基因进行单因素Cox回归分析,筛选<0.10的基因。为了进一步筛选重要的预后基因,避免单因素Cox回归分析的过度拟合问题,通过glmnet包[22]对单因素Cox回归分析结果中得到的基因进行500次迭代Lasso回归分析。选择被Lasso重复抽取大于400次的变量作为最终的关键预后基因[23]。
1.7 多因素Cox回归分析与预后模型构建
迭代Lasso回归分析筛选出的关键预后基因,基因迭代频次的大小反应了基因的重要程度。根据迭代频次大小排序,依次通过survival包纳入单个基因进行多因素Cox比例回归模型的构建,筛选接受者操作特性曲线(receiver operating characteristic curve, ROC曲线)下面积最大的基因集作为最佳预后模型。该步骤通过timeROC包[24]来完成。并用predict函数预测每个患者风险得分,将风险得分的中位数进行划分高低风险组进行KM生存分析。并用GEO、CGGA和Rembrandt数据库3个外部数据集来验证模型的优良。
1.8 富集分析
利用cluster profiler包[25]对短生存期组上调基因进行GO功能富集和KEGG通路富集分析,以确定在3个类别如生物学过程(biological process, BP)、细胞成分(cellular component, CC)和分子功能(molecular function, MF)中过度表达的功能富集以及过度表达的KEGG通路。对于该分析,FDR<0.05被认为具有统计学意义。基因富集分析(gene set enrichment analysis, GSEA)以不同生存期的差异分析后所得到的差异基因的Log2(fold change)值排序为输入文件,使用分子特征数据集(molecular signatures database, MSigDB)中GO基因集(C5),KEGG基因集(C2)和特征基因集(HALLMARK)注释基因富集情况。GSEA通过fgsea包[26]完成,标准化富集得分(normalized enrichment score, NES)和校正值用来评价基因集富集效果,其中校正<0.05为有统计学意义。
1.9 加权基因共表达网络构建
基于TCGA中GBM的RNA-seq表达谱数据,筛选出方差最大的前5000个基因,利用加权基因共表达网络(weighted gene co-expression network analysis, WGCNA)包[27]对这5000个基因的表达矩阵进行分析。WGCNA简要步骤如下:①基因模块识别:选择合适的软阈值β构建网络,最佳软阈值需达到无尺度拟合指数0.9以上,且需要网络的平均连通性较高。以此软阈值构建拓扑重叠矩阵(topological overlap matrix, TOM)。WGCNA将具有高拓扑重叠指数的一组基因定义为同一模块,对基因进行聚类,并按照混合动态剪切树的方法确定基因模块。②模块筛选:将模块进行主成分分析,即各模块第一主成分与临床表型进行皮尔森相关分析,得到模块与表型的相关系数即模块显著性(module significance, MS)及值,<0.05被认为有统计学意义。
我们运用WGCNA包构建了高低风险组的加权基因共表达网络。WGCNA具体参数如下:当使用0.9作为相关系数阈值时,我们选择的软阈值能力是4,并且选择10作为模块中的最小基因数。为了合并可能的相似模块,我们将0.2定义为切割高度的阈值。
1.10 统计分析
利用R 3.5.3软件进行数据下载、统计分析和相应的数据可视化。包括TCGAbiolinks包下载TCGA中GBM的RNA-seq表达谱数据、CNV数据和临床数据。利用Deseq2包对不同生存期进行差异分析。通过survival包进行KM生存分析、单变量、多变量Cox比例风险分析和模型构建,并利用survminer包进行相应的可视化。通过glmnet包进行迭代Lasso回归分析。利用ggplot2包和pheatmap包绘制风险因子关联图。
2 结果与分析
2.1 GBM不同生存期患者表达谱差异分析
用Deseq2包对TCGA中139例GBM的短生存期组(OS.time<1 year) 79例和长生存期组(OS.time> 3 year) 7例患者的表达谱数据进行差异分析,根据筛选标准<0.05且绝对值Log2(fold change)>1,我们一共筛选得到491个mRNA基因(附表1),其中上调基因426个,下调基因65个(图2)。为了探索短生存期组上调基因参与的生物学过程,进行了GO和KEGG富集分析。根据GO分析结果显示上调基因主要分布于细胞外基质、含胶原蛋白的细胞外基质、内质网内腔等组织;参与了细胞外组织、白细胞迁移、胶原组织等生物过程;主要与受体结合、生长因子结合等生物过程有关(图3A)。与上调基因相关的KEGG通路,包括细胞因子–受体相互作用,PI3K-Akt信号通路,肿瘤中的蛋白多糖,IL-17信号通路,肿瘤坏死因子信号通路等(图3B)。
为了弥补由于表达量阈值的影响,可能会遗漏某些差异的富集通路。我们对不同生存期差异分析的差异基因进行GSEA分析,按照校正后值从大到小排列,在GO基因集中,短生存期组显著富集了与细胞外基质、含胶原的细胞外基质和细胞外结构等生物过程(附图1A)。在KEGG基因集中,短生存期组显著富集了趋化因子信号通路、细胞因子受体相互作用等,与单独选取上调基因富集分析结果部分重叠(附图1B)。在HALLMARK基因集中,短生存期组显著富集了多种肿瘤相关的信号通路。如上皮间质型转化(epithelial-mesenchymal transition, EMT)、TNFA,KRSA信号通路、缺氧等(附图1C)。通过对不同生存期组间差异表达基因的GSEA分析,进一步确认了上述在短生存期组上调基因富集分析相关的信号通路和生物过程。
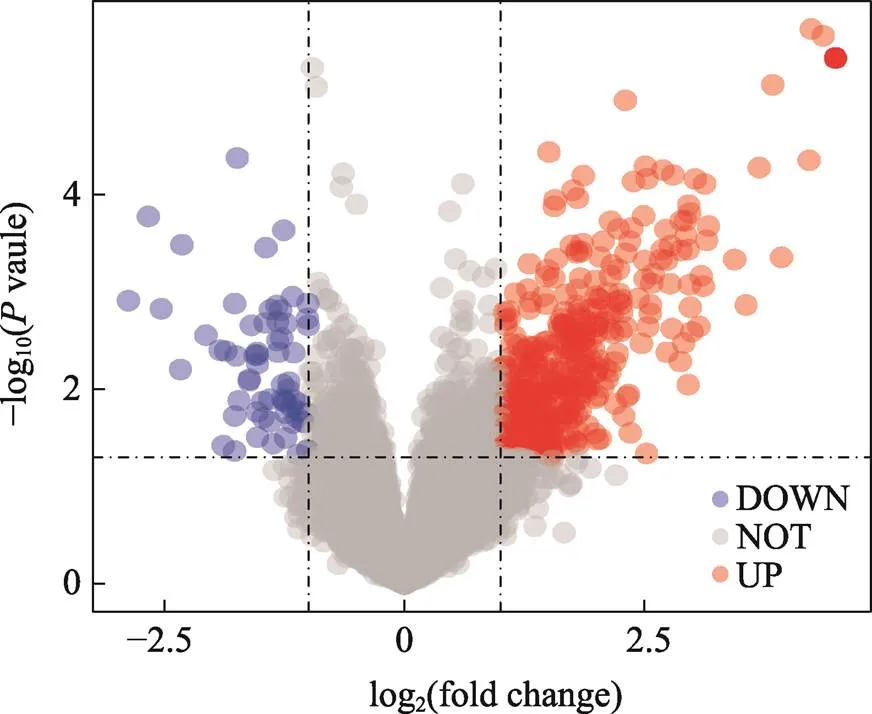
图2 TCGA中GBM患者不同生存期基因差异表达分析
差异基因火山图分析,红点代表上调的差异基因,蓝点代表下调的差异基因,灰点代表没有差异的基因。Log2(fold change)为差异分析中基因的差异倍数,OS.time<1 year比OS.time>3 year。
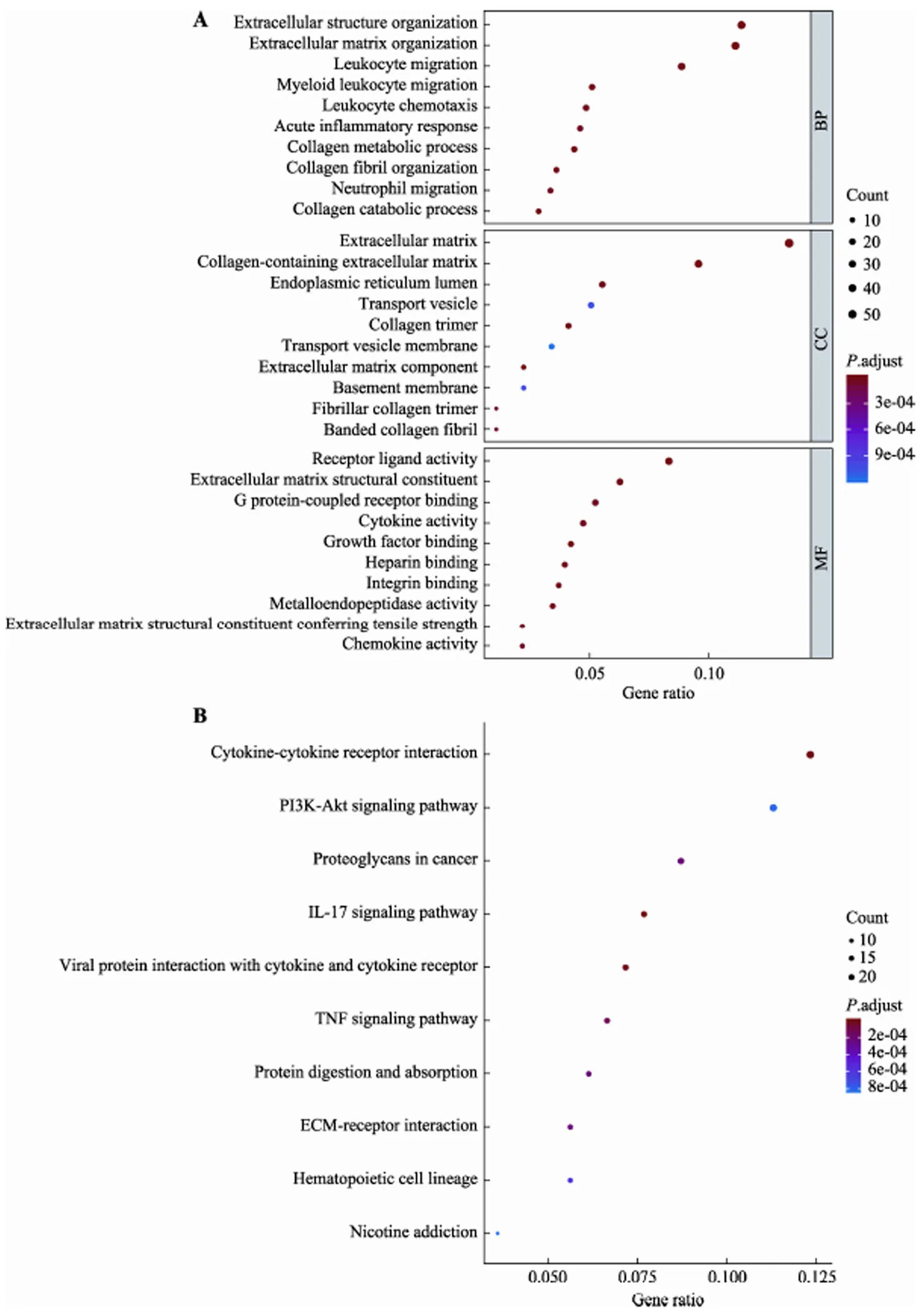
图3 TCGA中GBM短生存期组上调基因GO和KEGG富集分析
A:GO功能富集分析结果。BP:生物学过程;CC:细胞组分;MF:分子功能。B:KEGG通路富集分析结果。
2.2 GISTIC和KM分析CNV数据
对139例GBM的CNV数据进行GISTIC分析,得到所有基因离散化的拷贝数状态。将所有基因分成扩增组和非扩增组进行KM生存分析,根据<0.05得到2405个SAG。将短生存期组上调基因和SAG交集得到的47个候选基因,用于后续分析(附表2)。
2.3 构建GBM预后风险模型
为了进一步筛选预后基因,我们将得到的47个候选基因,进行单因素Cox比例风险分析,筛选<0.10的基因。筛选得到18个基因见表1所示。对上述基因集进行迭代Lasso回归,进行500次迭代处理,筛选频次大于400的基因。获得12个基因依据频次大小分别纳入多因素Cox回归模型,计算每次纳入基因的ROC曲线下的面积。选取面积最大的基因集合作为TCGA实验组的最佳模型。最终,我们确定了六基因(、、、、、)组合,六基因组合在多因素Cox回归模型中ROC曲线下的面积最大为0.912 (图4,A和B)。用predict函数预测TCGA实验组每个患者风险得分,根据风险得分的中位值(1.116),我们将139例中69例纳入高风险组,70例纳入低风险组。从风险因子关联图中可以看到,高风险组的死亡人数明显更多,存活人数更少(图4C)。6个基因表达量在患者中的分布表明,高风险组患者的6个基因的表达量比低风险组的更高。并且六基因模型的风险评分分组的KM分析显示,高低风险组存在明显差异。高风险组预后较差,<0.001具有统计学意义(图4D)。
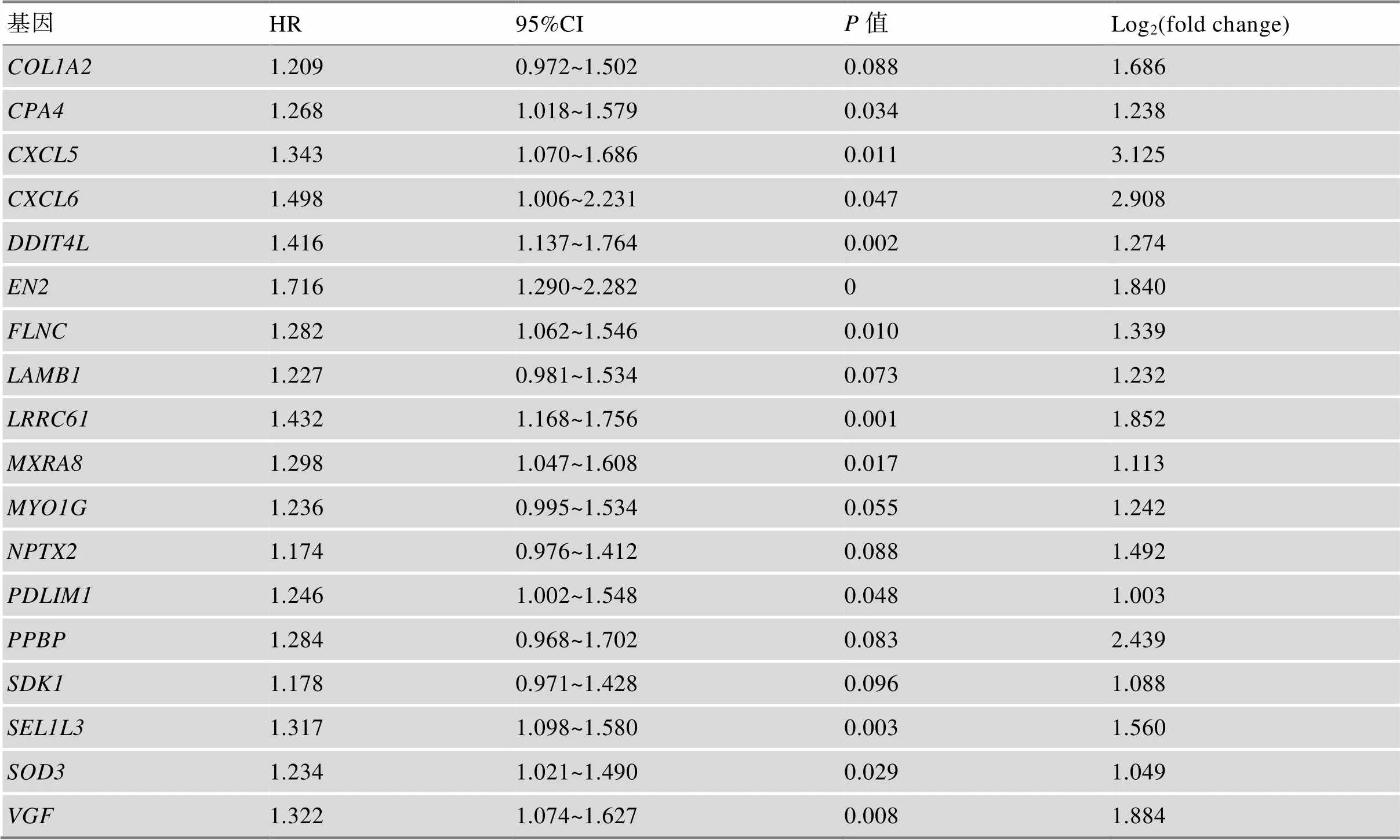
表1 单因素Cox回归筛选P<0.10的基因
HR为风险比(hazard ratio)用于描述相对危险度的指标。
Log2(fold change)为差异分析中基因的差异倍数,OS.time<1 year比OS.time>3 year。
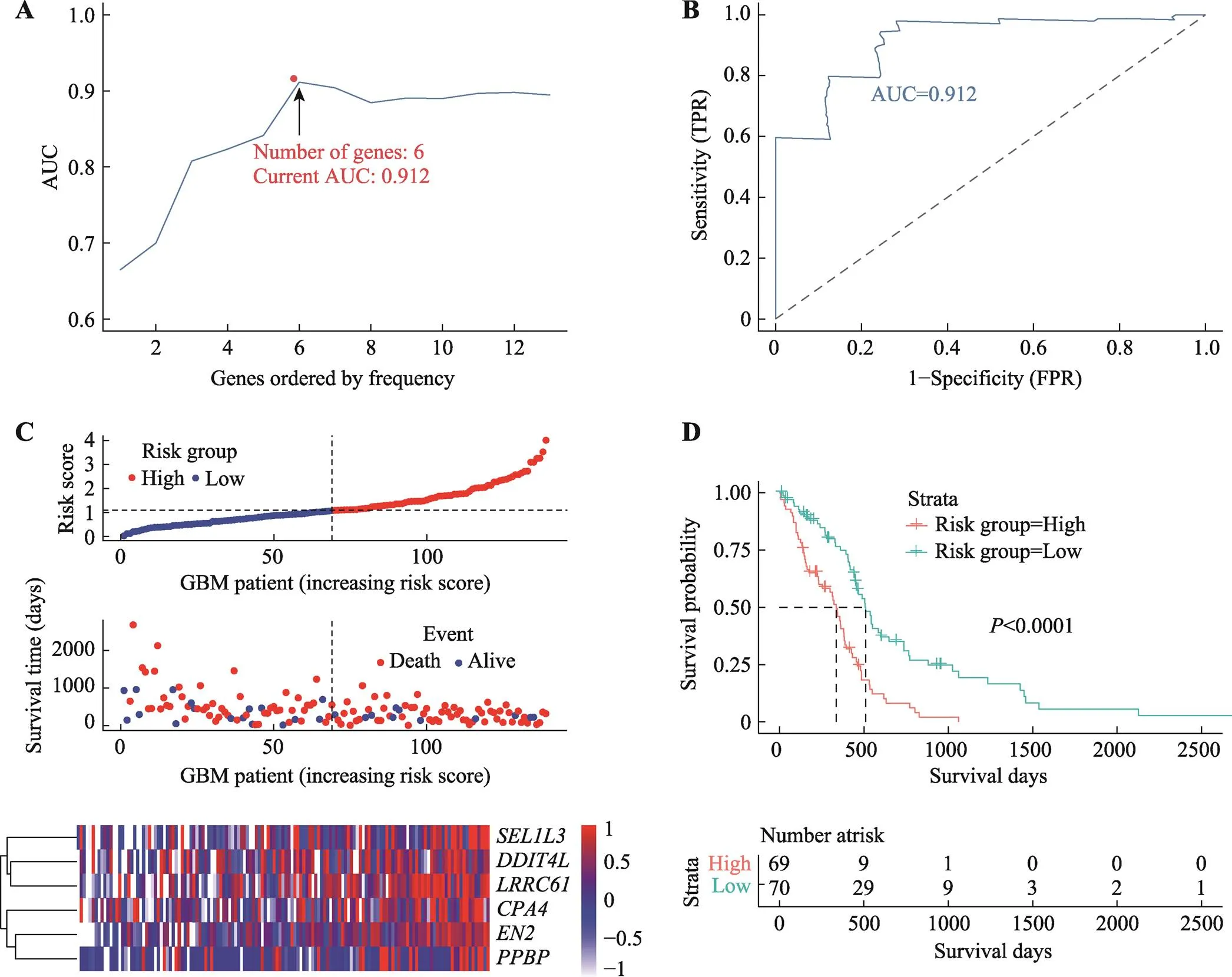
图4 TCGA中GBM实验组六基因预后模型建立
A:根据迭代Lasso回归的迭代次数纳入回归模型,选取曲线下面积的基因集合作为最佳模型。AUC (area under curve)被定义为ROC曲线下与坐标轴围成的面积。B:六基因预后模型的ROC曲线下面积为0.912。C:六基因预后模型风险因子关联图。上图:预后模型中高风险(红色)和低风险(蓝色) GBM患者的风险评分分布;中图:散点图显示了模型中GBM患者的生存状况。红点表示死亡的患者,蓝点表示存活的患者;下图:模型中6个基因的表达量热图,横坐标为基因表达量,纵坐标为GBM样本。D:高风险组和低风险组的KM生存分析。
2.4 外部数据验证六基因预后模型
为了对六基因预后模型进行验证,我们首先将六基因模型应用于GEO脑胶质瘤的GSE16011表达谱验证组,其ROC曲线下面积为0.825 (图5A)。说明六基因预后模型效能好,对于GBM的预后具有很好的预测能力。用predict函数预测GSE16011验证组每个患者风险得分,根据风险得分的中位值(1.0347),我们将150例中74例纳入高风险组,76例纳入低风险组。GSE16011验证组的风险因子关联图同TCGA实验组中具有一致性,高风险组的死亡人数多,存活人数少(图5B)。6个基因表达量随着风险评分增高而增高。KM生存分析同样显示了高风险组预后较差(图5C)。同样我们将六基因模型应用于CGGA验证组,结果显示,CGGA验证组的ROC曲线下面积为0.636 (附图2A),说明六基因预后模型在CGGA验证组有中等效能,对于GBM的预后具有一定的预测能力。CGGA验证组的风险因子关联图同TCGA实验组中具有一致性,高风险组的死亡人数多,存活人数少(附图2B)。用predict函数预测CGGA验证组每个患者风险得分,根据风险得分的中位值(0.998)我们将88例中44例纳入高风险组,44例纳入低风险组。CGGA验证组(附图2C) KM生存分析显示了高风险组预后较差的结果。进而我们将六基因模型应用于Rembrandt验证组,结果显示,其ROC曲线下面积为0.845 (附图3A),说明六基因预后模型效能好,对于GBM的预后具有很好的预测能力。Rembrandt验证组的风险因子关联图同TCGA实验组中具有一致性,高风险组的死亡人数多,存活人数少(附图3B)。用predict函数预测Rembrandt验证组每个患者风险得分,根据风险得分的中位值(0.986),我们将146例中73例纳入高风险组,73例纳入低风险组。Rembrandt验证组(附图3C) KM生存分析同样显示了高风险组预后较差的结果。值均小于0.05,具有统计学意义。从3个验证组结果看,六基因预后模型对实验组和外部数据验证组都有较好的预测作用,该模型可以对GBM患者的生存状态进行预测。
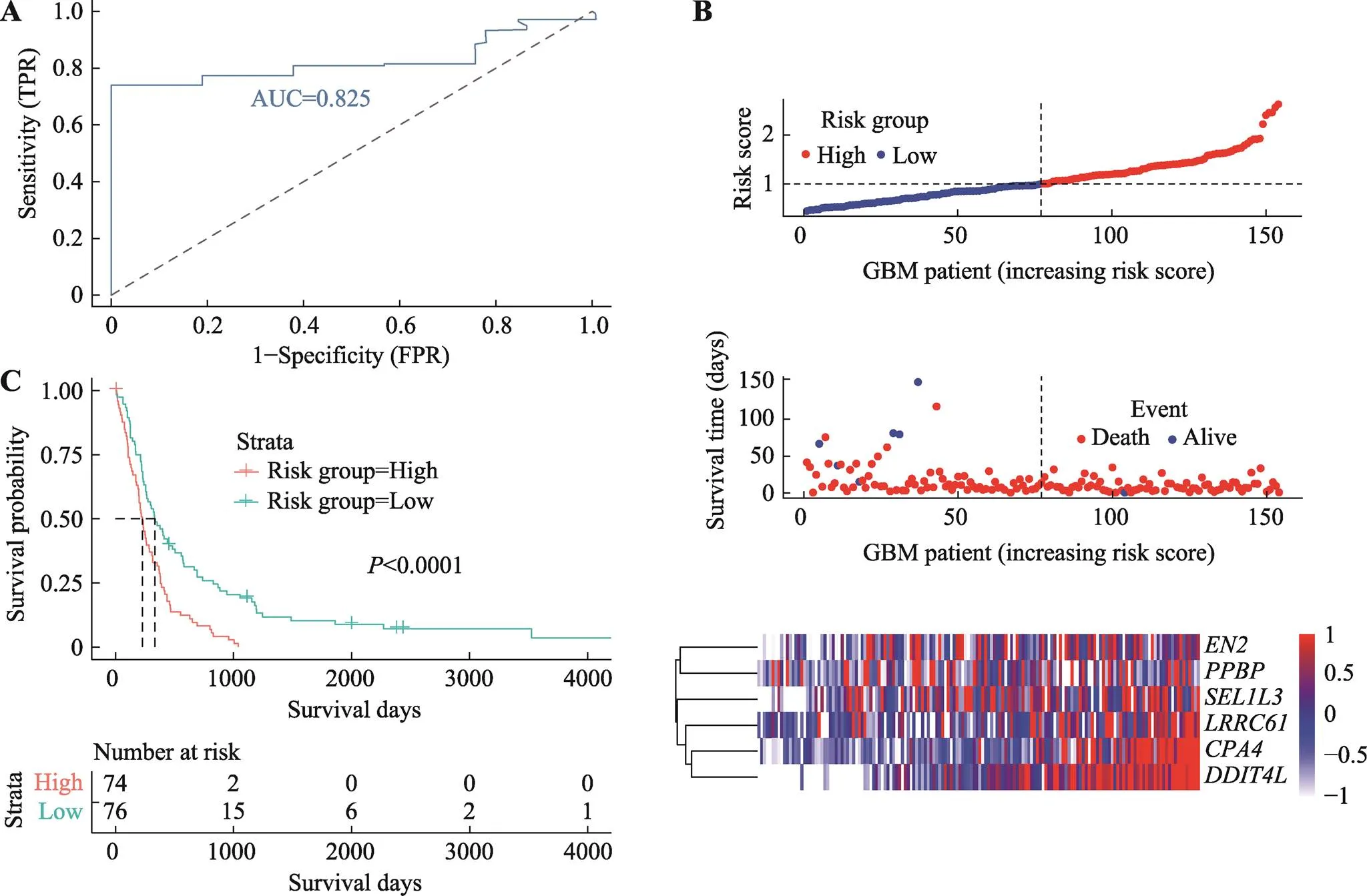
图5 GEO中GBM GSE16011验证组的六基因预后模型
A:六基因预后模型在GSE16011验证组的ROC曲线下面积为0.825。B:GSE16011验证组的六基因预后模型风险因子关联图。上图:预后模型中高风险(红色)和低风险(蓝色)GBM患者的风险评分分布;中图:散点图显示了模型中GBM患者的生存状况。红点表示死亡的患者,蓝点表示存活的患者;下图:模型中6个基因的表达量热图,横坐标为基因表达量,纵坐标为GBM样本。C:GSE16011验证组的高风险组和低风险组的KM生存分析。
2.5 评估六基因模型预后独立性
为了评估六基因模型的预后独立性,我们结合患者的临床指标包括:六基因预后评分分组、年龄、性别和基因突变状态等进行多因素Cox回归比例风险模型。结果见表2所示。在TCGA GBM实验组中,多因素Cox回归分析显示,六基因评分风险分组(HR=2.39, 95%CI=1.582~3.617,=0.0004)和年龄(HR=1.02, 95%CI=1.005~1.04,=0.01056)与生存显著相关,并且均为危险因素,HR均大于1。在GEO GSE16011验证组中,多因素Cox回归分析显示,六基因评分风险分组(HR=1.46, 95%CI=1.012~ 2.125,=0.04285)和年龄(HR=1.03, 95%CI=1.02~ 1.052,=0.0001)与生存显著相关,同样均为危险因素,HR大于1。在CGGA验证组中,多因素Cox回归分析显示,六基因评分风险分组(HR=1.92, 95%CI=1.189~3.102,=0.00766)与生存显著相关,为危险因素,HR大于1。在Rembrandt验证组中,多因素Cox回归分析显示,六基因评分风险分组(HR=1.76, 95%CI=1.18~2.61,=0.005)与生存显著相关,为危险因素,HR大于1。综上所述,六基因预后模型可以作为一个独立于其他临床因素的预后指标,用于GBM的风险预后。

表2 多因素Cox回归比例风险模型
2.6 加权共表达网络分析
通过WGCNA包将RNA-seq表达谱数据中方差最大的前5000基因用于加权共表达网络分析。当使用0.9作为相关系数阈值时,软阈值被选为4(图6A)。通过WGCNA分析,构建了13个共表达模块(图6B),与性状相关分析显示黑色模块与高风险组最相关,模块显著性(MS)为0.33,<0.05 (图6C)。我们对黑色模块包含的206个基因进行GO功能和KEGG富集分析。GO功能富集分析结果显示,模块内的基因主要分布在细胞外基质,含胶原蛋白的细胞外基质,内质网内腔等组织,参与了细胞外组织、白细胞迁移、与氧反应等生物过程;主要有与受体相关,生长因子相关,胞外基质结合等分子功能(图7A)。与模块内的基因相关的KEGG通路,包括细胞因子–细胞因子受体相互作用、肿瘤坏死因子信号通路、焦点粘着、类风湿性关节炎、IL−17信号通路等(图7B)。与前面的上调基因富集分析在GO功能和KEGG通路上具有高度一致性。
3 讨论
GBM作为最常见、恶性程度最高的胶质瘤亚型,虽然总体预后差,但是具有广泛分子异质性和患者的生存期差异大的特点。目前研究表明可以作为预测GBM预后的相关分子生物标志物,包括基因突变、染色体1p/19q联合缺失和基因启动子甲基化等。具有上述相关分子改变的GBM患者对化疗敏感并且具有预后较好的特点。然而具有扩增、重排、、突变和融合或点突变的分子改变患者提示预后不佳的效果。实际上,目前已有研究通过筛选一系列与GBM相关的功能核心基因来建立预后模型,如自噬、缺氧、免疫等方面相关分子。上述特征是基于单一转录组学数据的生物学过程发展起来的。一个层面数据的改变很难解释疾病的整个发生过程。因此,本研究对TCGA数据库中GBM患者的RNA-seq表达谱数据和CNV数据联合分析,建立六基因(、、、、、)预后模型,发现TCGA GBM实验组中该风险模型是很好的预后预测因子,能够很好地识别预后不良高风险的GBM患者;并且在GEO中GSE16011验证组、CGGA验证组和Rembrandt验证组显示该模型具有良好的稳定性和可重复性。相较与以往研究,多组学联合分析更有利于全面评估患者的预后风险,同时也为后续的分析研究提供了思路。基于基因组和转录组构建的预测模型能够很好地识别预后不良高风险的GBM患者,并且能够在不同数据集中得到很好的验证,结果具有一致性,故能为GBM患者的预后风险评估提供更好的依据。
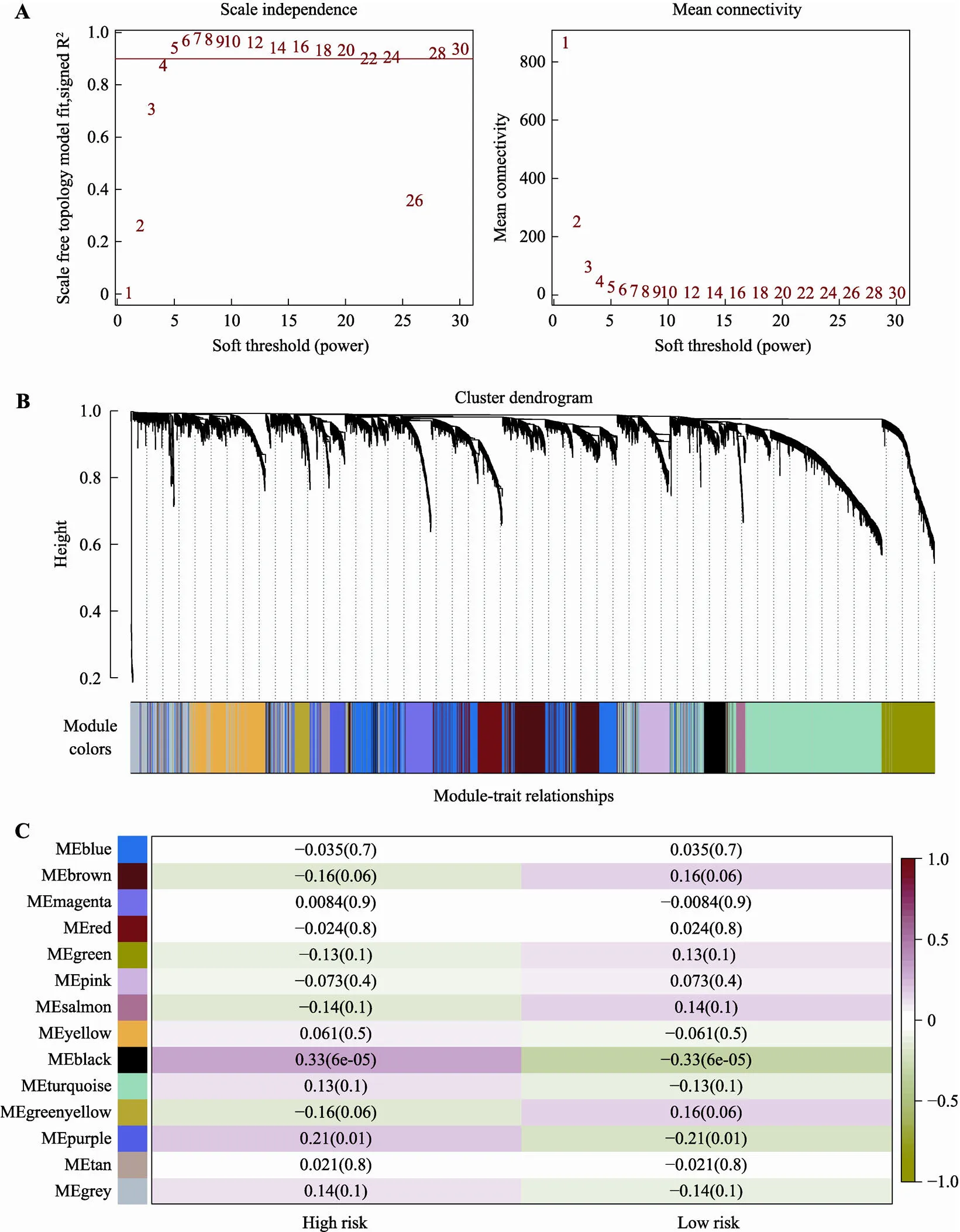
图6 WGCNA加权共表达网络识别高风险组相关模块
A:基于TCGA中GBM RNA-seq表达数据的软阈值的确定。B:基因聚类树状图。树形图下方的色块表示由动态树切割方法识别的模块,模块内的基因高度相关。C:模块与高低风险组相关性的热图。色块中上面的数字代表相关性,下面数字代表值,红色呈正相关,绿色呈负相关。
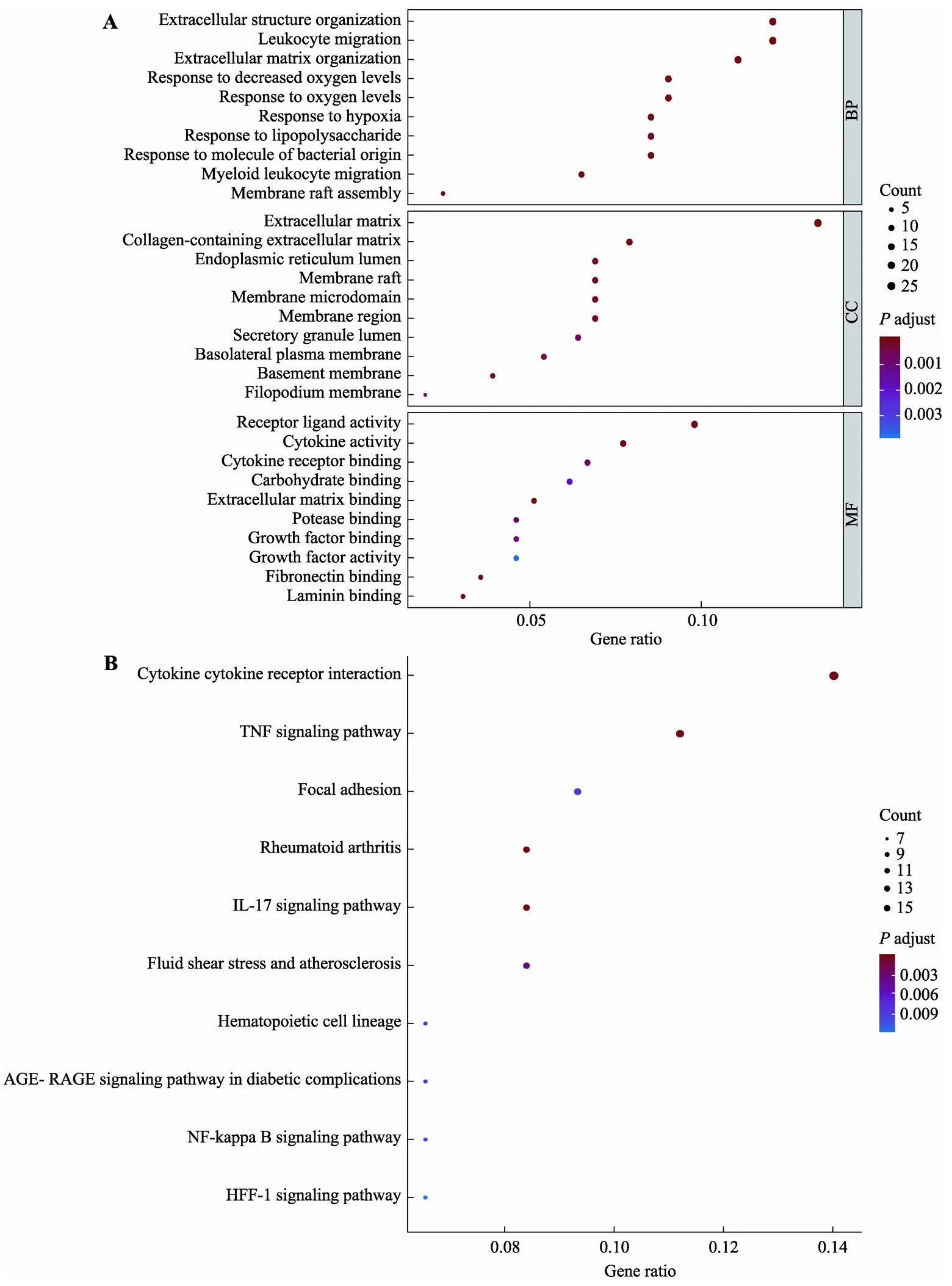
图7 高风险组相关模块内的基因GO和KEGG富集分析
A:高风险组相关模块内的基因GO功能富集分析结果。BP:生物学过程;CC:细胞组分;MF:分子功能。B:高风险组相关模块内的基因通路富集分析结果。
六基因模型区分的高风险组相关基因的富集分析结果,与上调基因的富集分析结果核心成分相重叠,主要集中在细胞外基质、白细胞迁移、细胞外组织、受体结合和生长因子相关等功能;细胞因子受体相互作用,肿瘤坏死因子信号通路,IL−17信号通路等。并且在GSEA分析HALLMARK基因集中,短生存期组显著富集了肿瘤EMT信号通路,缺氧等其他经典肿瘤相关信号通路。细胞外基质相关生物过程通常与肿瘤发生EMT甚至侵袭迁移的恶性表型密切相关[28]。因此,六基因模型可以获得与GBM预后相关的核心细胞生物学过程,细胞组分,分子功能及相关通路。进一步说明了六基因组合模型可以较好地用于预测GBM患者的预后。
在胶质瘤以往的研究报道中,6个基因中有4个基因与胶质瘤密切相关。基因编码一种含有同源结构域的转录因子,在胚胎神经发育过程中是必不可少的。LEF1-AS1通过上调EN2参与GBM的恶性发展[29]。基因编码的蛋白质是一种血小板衍生生长因子,属于CXC趋化因子家族,这种生长因子是一种有效的中性粒细胞趋化剂和激活剂。它被证明可以刺激各种细胞过程,包括DNA合成、有丝分裂、糖酵解等,在发育和治疗耐药中发挥作用。PPBP趋化因子可能是未来GBM研究的一个有希望的靶点[30],该基因编码的蛋白也被预测作为肺癌早期诊断的标志物[31]。基因编码富含亮氨酸重复序列蛋白61。在人类蛋白质图谱(Human Protein Atlas, HPA, https://www.proteinatlas. org/)数据库中,免疫组化结果显示LRRC61在高级别胶质瘤组织和低级别胶质瘤组织中表达高于正常大脑皮层组织(附图4,A~C)。基因编码SEL1L家族成员3,在HPA数据库中,免疫组化结果显示SEL1L3也在高级别胶质瘤组织和低级别胶质瘤组织中表达高于正常大脑皮层组织(附图4,D~F)。另外两种基因中,基因是羧肽酶A/B亚家族的一员,与第7号染色体上的另外3个家族成员位于一个簇中。羧肽酶是一类含锌的外肽酶,催化羧基末端氨基酸的释放,并被合成为酶原,通过蛋白水解被激活。该基因可能参与组蛋白高乙酰化途径[32],有文献报道,通过CPA4激活STAT3和ERK1/2信号通路促进细胞生长并预测结直肠癌预后不良[33]。基因编码DNA损伤诱导转录样蛋白4,有研究表明DDIT4L可能是通过心脏mTOR信号传导病理应激到自噬的重要传感器,并且DDIT4L可以作为心血管疾病的治疗靶点,在自噬和mTOR信号通路起主要作用[34]。
综上所述,根据我们的研究,提出六基因(、、、、、)预后模型可为GBM患者的预后风险评估提供更好的依据。其包含已知与GBM相关的基因和未知基因,值得我们在今后的临床样本中验证,并进行更多的临床学与功能学研究。
附录:
附加材料详见文章电子版www.chinagene.cn。
致谢:
感谢哈尔滨医科大学傅松滨教授在指导文章写作和审阅文章过程中给予的帮助。
[1] Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma., 2006, 2(9): 494–503.
[2] Su CL, Li L, Chen XW, Zhang J, Shen NX, Wang ZX, Yang SQ, Li J, Zhu WZ, Wang CY. Summary of classification of central nervous system tumors in WHO 2016., 2016, 31(7): 570–579.苏昌亮, 李丽, 陈小伟, 张巨, 申楠茜, 王振熊, 杨时骐, 李娟, 朱文珍, 王承缘. 2016年WHO中枢神经系统肿瘤分类总结. 放射学实践, 2016, 31(7): 570–579.
[3] Parsons DW, Jones S, Zhang XS, Lin JCH, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Hartigan J, Smith DR, Strausberg RL, Marie SKN, Shinjo SMO, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme., 2008, 321(5897): 1807–1812.
[4] Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, Sabel M, Steinbach JP, Heese O, Reifenberger G, Weller M, Schackert G, German Glioma Network. Long-term survival with glioblastoma multiforme., 2007, 130: 2596–2606.
[5] Gerber NK, Goenka A, Turcan S, Reyngold M, Makarov V, Kannan K, Beal K, Omuro A, Yamada Y, Gutin P, Brennan CW, Huse JT, Chan TA. Transcriptional diversity of long-term glioblastoma survivors., 2014, 16(9): 1186–1195.
[6] Asiedu MK, Thomas CF, Dong J, Schulte SC, Khadka P, Sun ZF, Kosari F, Jen J, Molina J, Vasmatzis G, Kuang R, Aubry MC, Yang P, Wigle DA. Pathways impacted by genomic alterations in pulmonary carcinoid tumors., 2018, 24(7): 1691–1704.
[7] Chang J, Tan WL, Ling ZQ, Xi RB, Shao MM, Chen MJ, Luo YY, Zhao YJ, Liu Y, Huang XC, Xia YC, Hu JL, Parker JS, Marron D, Cui QH, Peng LN, Chu JH, Li HM, Du ZL, Han YL, Tan W, Liu ZH, Zhan QM, Li Y, Mao WM, Wu C, Lin DX. Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking- related mutation signature and genomic alterations., 2017, 8: 15290.
[8] Liang L, Fang JY, Xu J. Gastric cancer and gene copy number variation: emerging cancer drivers for targeted therapy., 2016, 35(12): 1475–1482.
[9] Wang Z, Wang LX. Gene amplification in lung cancer., 2010, 33(3): 155–159.王箴, 王良旭. 基因扩增在肺癌中的研究进展. 国际遗传学杂志, 2010, 33(3): 155–159.
[10] Ohgaki H, Kleihues P. Genetic alterations and signaling pathways in the evolution of gliomas., 2009, 100(12): 2235–2241.
[11] Jin WX, Yang TM, Dong XY, Hua ZC, Xu XX. P53 mutation in human brain gliomas: Comparison of loss of heterozygosity with mutation frequency., 2000, 22(6): 357–360.金卫新, 杨天明, 董雪吟, 华子春, 徐贤秀. 脑胶质瘤中P53基因突变及其与染色体17p杂合性丢失的比较. 遗传, 2000, 22(6): 357–360.
[12] Wang YL, Zhao WJ, Xiao Z, Guan GF, Liu X, Zhuang MH. A risk signature with four autophagy-related genes for predicting survival of glioblastoma multiforme., 2020, 24(7): 3807–3821.
[13] Lin WZ, Wu SH, Chen XC, Ye YL, Weng YL, Pan YH, Chen ZJ, Chen L, Qiu XX, Qiu SF. Characterization of hypoxia signature to evaluate the tumor immune microenvironment and predict prognosis in glioma groups., 2020, 10: 796.
[14] Wang JJ, Wang H, Zhu BL, Wang X, Qian YH, Xie L, Wang WJ, Zhu J, Chen XY, Wang JM, Ding ZL. Development of a prognostic model of glioma based on immune-related genes., 2021, 21(2): 116.
[15] Hermansen SK, Sørensen MD, Hansen A, Knudsen S, Alvarado AG, Lathia JD, Kristensen BW. A 4-miRNA signature to predict survival in glioblastomas., 2017, 12(11): e0188090.
[16] Jia DY, Lin W, Tang HL, Cheng YF, Xu KW, He YS, Geng WJ, Dai QX. Integrative analysis of DNA methylation and gene expression to identify key epigenetic genes in glioblastoma., 2019, 11(15): 5579– 5592.
[17] Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, Ceccarelli M, Bontempi G, Noushmehr H. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data., 2016, 44(8): e71.
[18] Li X, Li MW, Zhang YN, Xu HM. Common cancer genetic analysis methods and application study based on TCGA database., 2019, 41(3): 234–242.李鑫, 李梦玮, 张依楠, 徐寒梅. 常用肿瘤基因分析方法及基于TCGA数据库的分析应用. 遗传, 2019, 41(3): 234–242.
[19] Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2., 2014, 15(12): 550.
[20] Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, Vivanco I, Lee JC, Huang JH, Alexander S, Du JY, Kau T, Thomas RK, Shah K, Soto H, Perner S, Prensner J, Debiasi RM, Demichelis F, Hatton C, Rubin MA, Garraway LA, Nelson SF, Liau L, Mischel PS, Cloughesy TF, Meyerson M, Golub TA, Lander ES, Mellinghoff I, Sellers WR. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma., 2007, 104(50): 20007–20012.
[21] Stalpers LJA, Kaplan EL. Edward L. Kaplan and the Kaplan-Meier survival curve., 2018, 33(2): 109–135.
[22] Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent., 2010, 33(1): 1–22.
[23] Sveen A, Ågesen TH, Nesbakken A, Meling GI, Rognum TO, Liestøl K, Skotheim RI, Lothe RA. ColoGuidePro: a prognostic 7-gene expression signature for stage III colorectal cancer patients., 2012, 18(21): 6001–6010.
[24] Blanche P, Dartigues JF, Jacqmin-Gadda H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks., 2013, 32(30): 5381–5397.
[25] Yu GC, Wang LG, Han YY, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters., 2012, 16(5): 284–287.
[26] Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov M, Sergushichev A. Fast gene set enrichment analysis., 2019: 060012.
[27] Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis., 2008, 9: 559.
[28] Zeng Y, Xiao HL, Guo QN. Research progress on the mechanism of epithelial mesenchymal transition in glioblastoma., 2019, 26(8): 533–538.曾英, 肖华亮, 郭乔楠. 胶质母细胞瘤上皮间叶转化机制研究进展. 诊断病理学杂志, 2019, 26(8): 533–538.
[29] Zeng S, Zhou C, Yang DH, Xu LS, Yang HJ, Xu MH, Wang H. LEF1-AS1 is implicated in the malignant development of glioblastoma via sponging miR-543 to upregulate EN2., 2020, 1736: 146781.
[30] Achyut BR, Shankar A, Iskander ASM, Ara R, Angara K, Zeng P, Knight RA, Scicli AG, Arbab AS. Bone marrow derived myeloid cells orchestrate antiangiogenic resistance in glioblastoma through coordinated molecular networks., 2015, 369(2): 416–426.
[31] Du Q, Li EC, Liu YE, Xie WL, Huang C, Song JQ, Zhang W, Zheng YJ, Wang HL, Wang Q. CTAPIII/CXCL7: a novel biomarker for early diagnosis of lung cancer., 2018, 7(2): 325–335.
[32] Ross PL, Cheng I, Liu X, Cicek MS, Carroll PR, Casey G, Witte JS. Carboxypeptidase 4 gene variants and early-onset intermediate-to-high risk prostate cancer., 2009, 9: 69.
[33] Pan HD, Pan JX, Ji L, Song SB, Lv H, Yang ZR, Guo YB. Carboxypeptidase A4 promotes cell growth via activating STAT3 and ERK signaling pathways and predicts a poor prognosis in colorectal cancer., 2019, 138: 125–134.
[34] Simonson B, Subramanya V, Chan MC, Zhang AF, Franchino H, Ottaviano F, Mishra MK, Knight AC, Hunt D, Ghiran I, Khurana TS, Kontaridis MI, Rosenzweig A, Das S. DDiT4L promotes autophagy and inhibits pathological cardiac hypertrophy in response to stress., 2017, 10(468): eaaf5967.
附录

附图1 TCGA中GBM不同生存期差异基因GSEA分析结果
Supplementary Fig. 1 GSEA analysis genes of GBM patients with different survival time in TCGA
A:GSEA分析GO基因集中,短生存期组显著富集的结果。B:GSEA分析KEGG基因集中,短生存期组和长生存期显著富集的结果。C:GSEA分析HALLMARK基因集中,短生存期组和长生存期显著富集的结果。NES为normalized enrichment score,标准化富集得分。
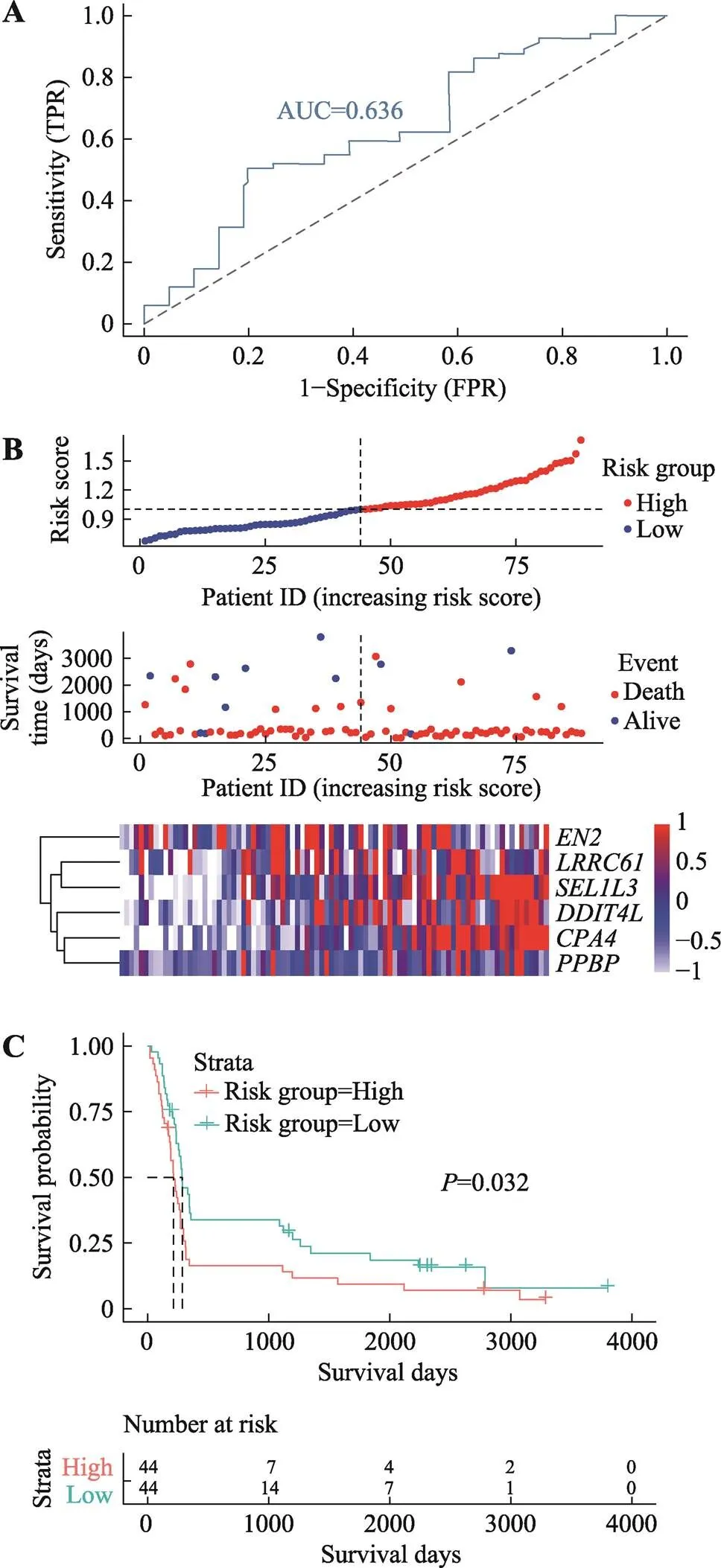
附图2 CGGA验证组的六基因预后模型
Supplementary Fig. 2 Six-gene prognostic model of CGGA validation group
A:六基因预后模型在CGGA验证组的ROC曲线下面积为0.636。B:CGGA验证组的六基因预后模型风险因子关联图。上图为预后模型中高风险(红色)和低风险(蓝色) GBM患者的风险评分分布;中图:散点图显示了模型中GBM患者的生存状况。红点表示死亡的患者,蓝点表示存活的患者;下图为模型中6个基因的表达量热图,横坐标为基因表达量,纵坐标为GBM样本。C:CGGA验证组的高风险组和低风险组的KM生存分析。
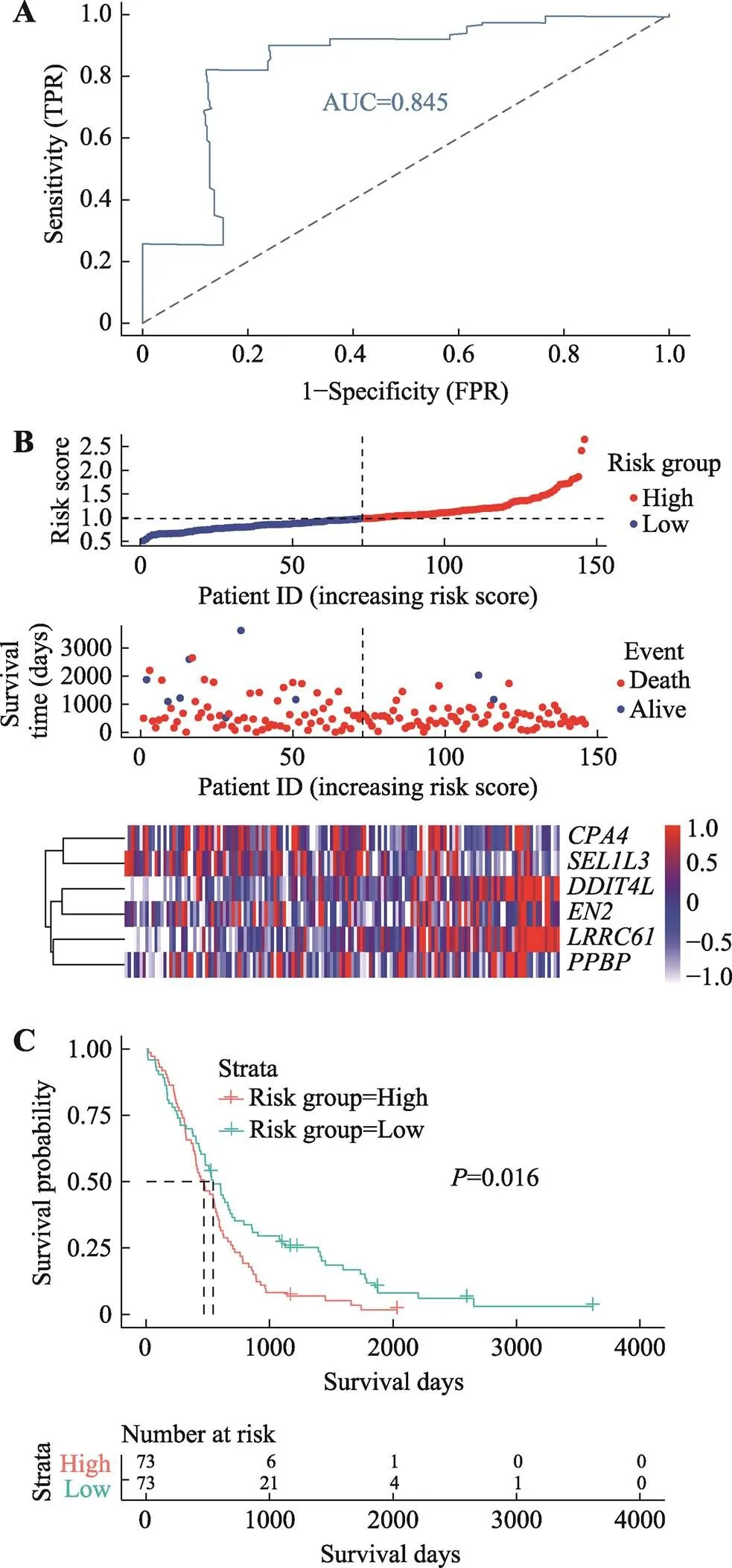
附图3 Rembrandt验证组的六基因预后模型
Supplementary Fig. 3 Six-gene prognostic model of Rembrandt validation group
A:六基因预后模型在Rembrandt验证组的ROC曲线下面积为0.845。B:Rembrandt验证组的六基因预后模型风险因子关联图。上图为预后模型中高风险(红色)和低风险(蓝色) GBM患者的风险评分分布;中图:散点图显示了模型中GBM患者的生存状况。红点表示死亡的患者,蓝点表示存活的患者;下图为模型中6个基因的表达量热图,横坐标为基因表达量,纵坐标为GBM样本。C:Rembrandt验证组的高风险组和低风险组的KM生存分析。
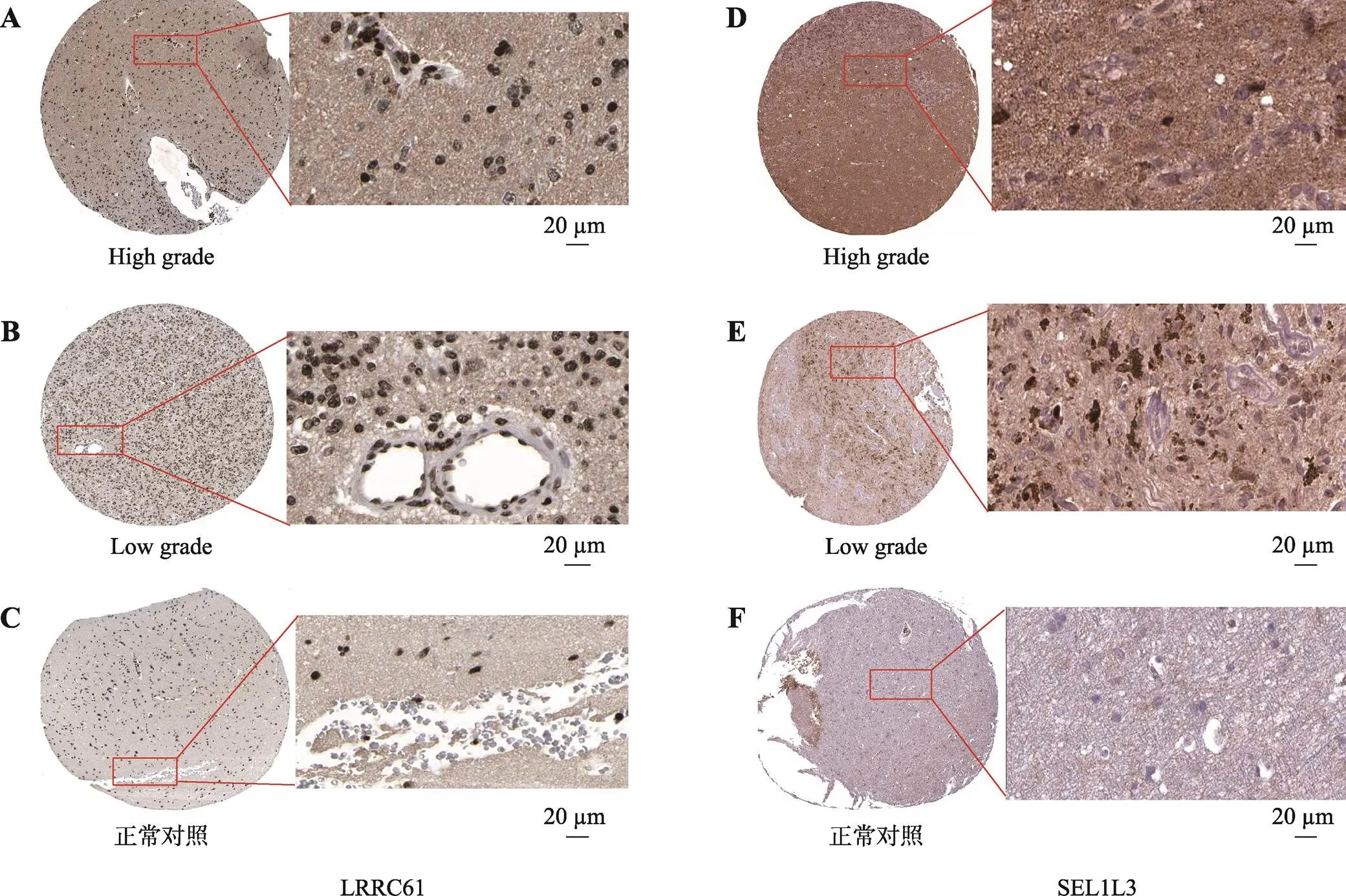
附图4 预后模型的基因在胶质瘤组织和正常组大脑皮层组织中的蛋白表达情况
Supplementary Fig. 4 Protein expression of prognostic model genes in glioma tissue
A:LRRC61在正常大脑皮层组织中的免疫组化结果;B:LRRC61在低级别胶质瘤组织中的免疫组化结果;C:LRRC61在高级别胶质瘤组织中的免疫组化结果;D:SEL1L3在在正常大脑皮层组织中的免疫组化结果;E:SEL1L3在低级别胶质瘤组织组织中的免疫组化结果;F:SEL1L3在高级别胶质瘤组织中的免疫组化结果,图片数据来源于人类蛋白质图谱数据库(HPA)。
附表1 TCGA中GBM患者不同生存期的差异基因
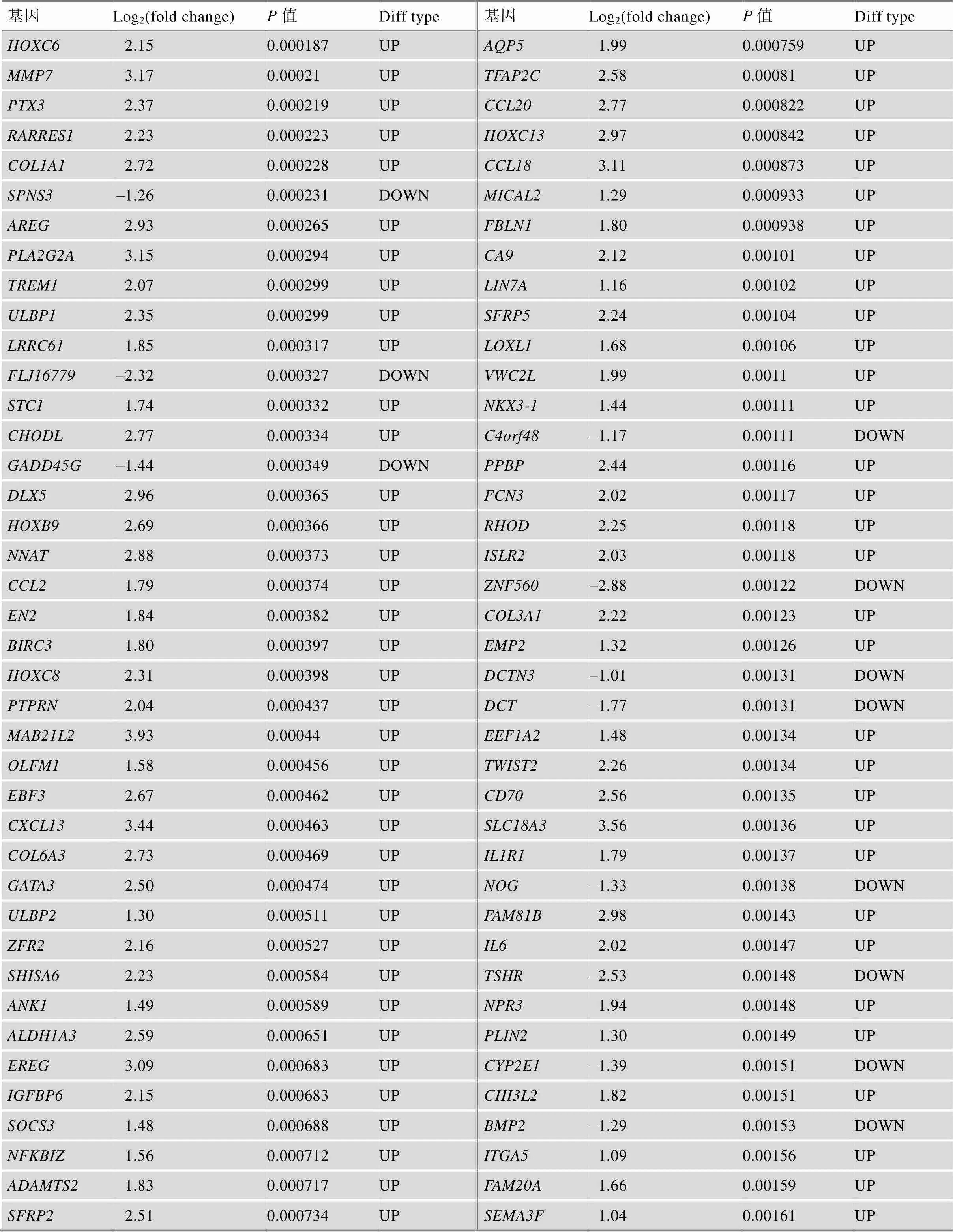
续附表1

续附表1

续附表1
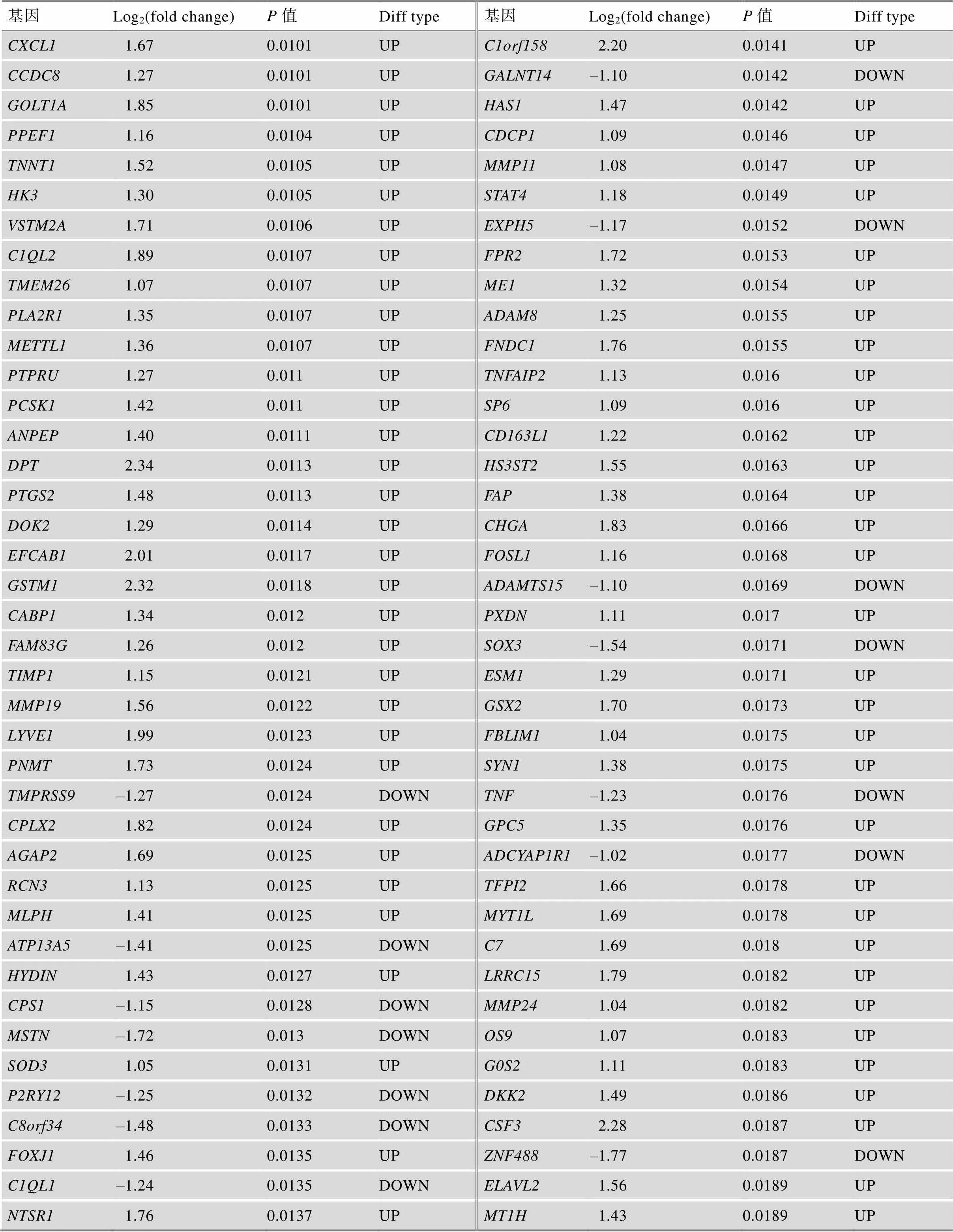
续附表1
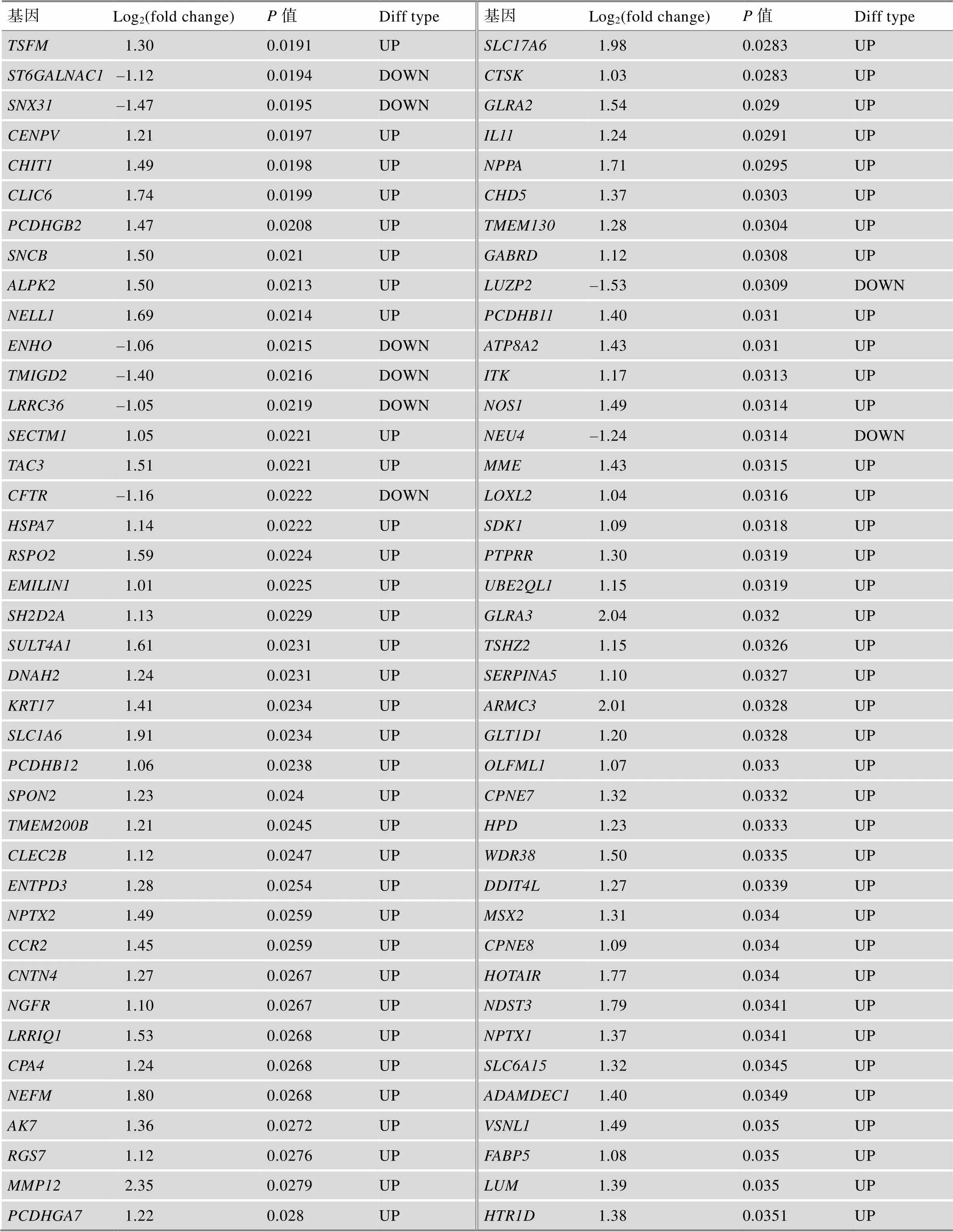
续附表1
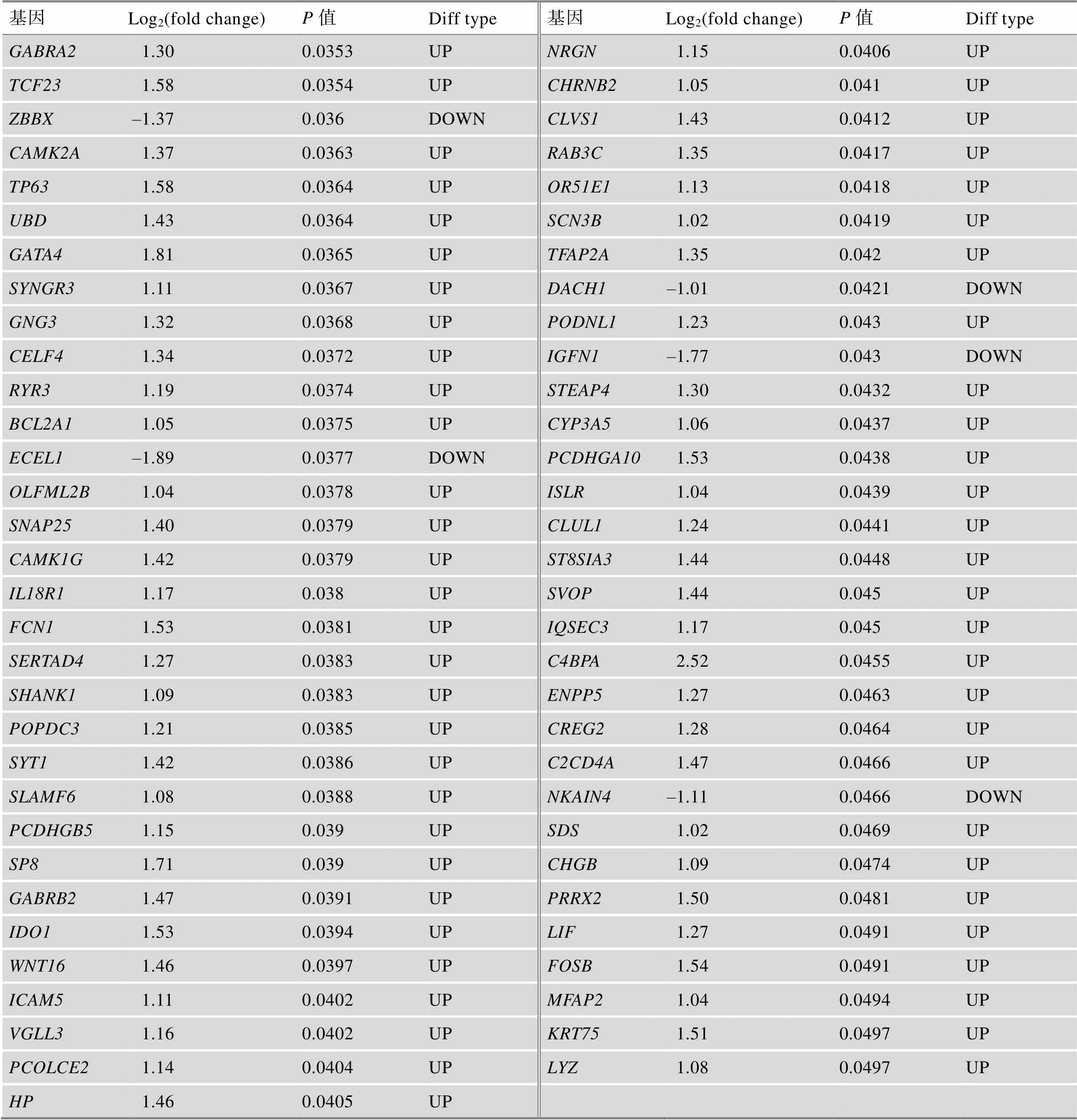
续附表1
Log2(fold change)为差异分析中基因的差异倍数,OS.time<1 year比OS.time>3 year。
Diff type为差异基因类型,UP为上调基因,DOWN为下调基因。
附表2 短生存期组上调基因和SAG交集结果
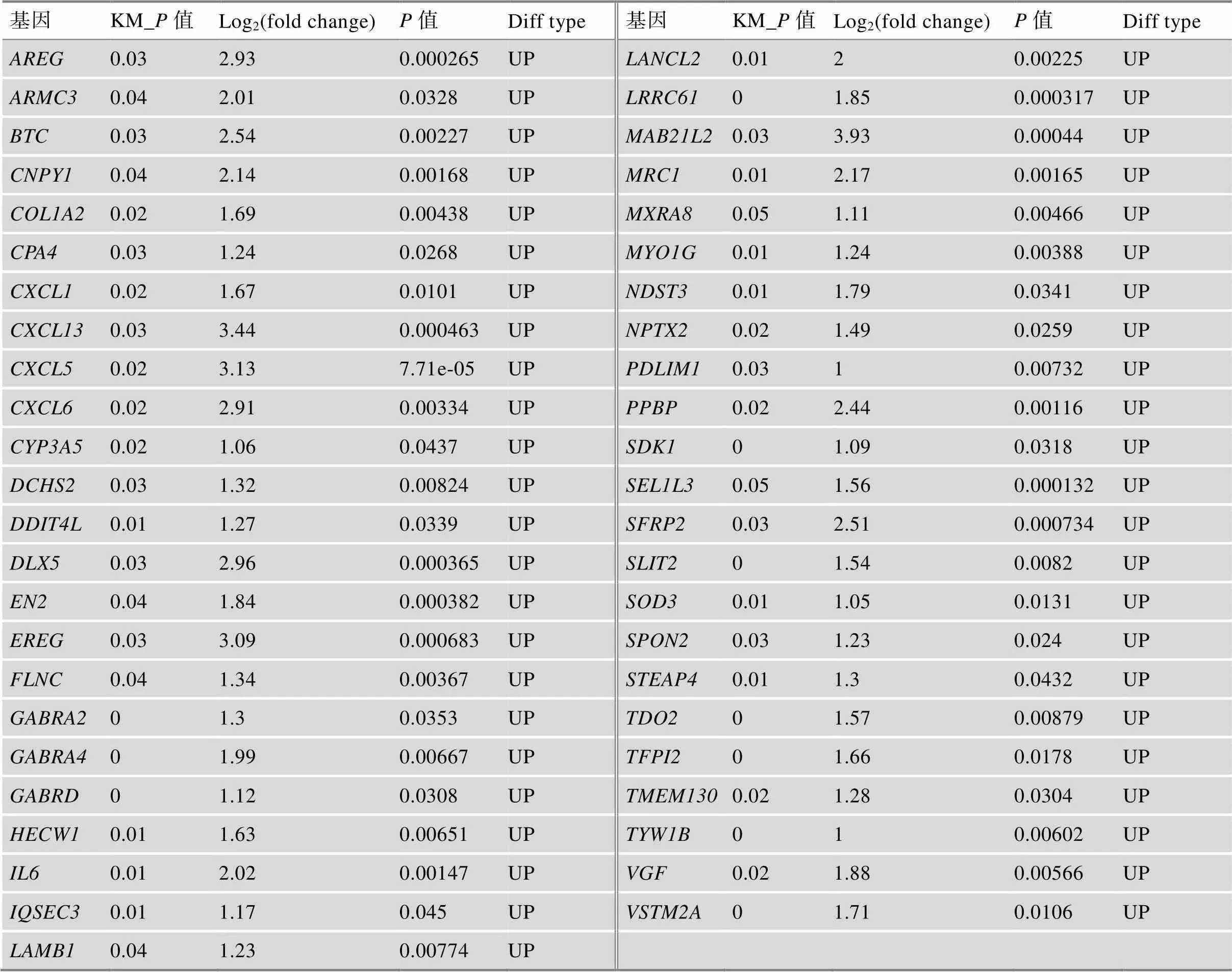
Supplementary Table 2 The intersection of up-regulated genes in short survival group and SAG
KM_为KM生存分析的值。为不同生存期组患者差异分析的值。
Establish six-gene prognostic model for glioblastoma based on multi-omics data of TCGA database
Changgui Lei, Xueyuan Jia, Wenjing Sun
Glioblastoma (GBM) is the most common primary intracranial tumor with extremely high malignancy and poor prognosis.In order to identify the GBM prognostic biomarkers and establish a prognostic model, we analyzed the expression profile data of GBM in The Cancer Genome Atlas (TCGA) database as the experimental group. First, we identified the differentially expressed genes of different survival periods among the GBM patients. The GISTIC software and Kaplan Meier (KM) survival curve were used to analyze the copy number variation of GBM to identify the survival-associated amplified gene (SAG). We selected the intersection genes of up-regulated ones in short survival group and SAG, performed univariate Cox regression and iterative Lasso regression with them to identify the important candidate genes and establish a prognostic model. Based on the model, the prognostic score was calculated. The patients were divided into high-risk and low-risk groups according to the median prognostic score. Meanwhile ROC curve was used to evaluate the validity of the model, applying the KM survival analysis of the high-risk and low-risk groups. Multivariate Cox regression analysis was used to determine the independence of the prognostic score. All the data were verified with three external datasets: GEO GSE16011, CGGA, and Rembrandt. The results showed that differential expression analysis of different survival periods of GBM identified 426 up-regulated genes and 65 down-regulated genes in the TCGA GBM dataset. The intersection of up-regulated genes in short survival group and SAG yielded 47 genes. After the screening, the six-gene combination (,,,,,) prognostic model was finally determined. The area under ROC curve of the model in TCGA experimental group and three external validation group were all greater than 0.6, even reaching 0.912. KM analysis showed that the prognosis of the high-risk and low-risk groups was significant different (<0.05). In the multivariate Cox regression analysis, the six-gene prognostic score was an independent factor influencing the prognosis of GBM patients (<0.05).In summary, this study established a prognostic model of six-gene (,,,,,) for GBM. This six-gene model has good predictive ability and could be used as an independent prognostic marker for GBM patients.
glioblastoma; multi-omics data; six-gene combination; prognostic model; The Cancer Genome Atlas
2020-12-11;
2021-03-20
教育部“创新团队发展计划”项目(编号IRT1230)资助[Supported by Program for Changjiang Scholars and Innovative Research Team in University (No. IRT1230)]
雷常贵,在读硕士研究生,专业方向:医学遗传学。E-mail: leichanggui@hrbmu.edu.cn
贾学渊,博士,副教授,研究方向:医学遗传学。E-mail: jiaxueyuan@hrbmu.edu.cn
雷常贵和贾学渊并列第一作者。
孙文靖,博士,教授,研究方向:医学遗传学。E-mail: sunwj@ems.hrbmu.edu.cn
10.16288/j.yczz.20-428
2021/4/7 16:51:07
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20210407.1059.002.html
(责任编委: 何顺民)
猜你喜欢
核安全(2022年2期)2022-05-05
中国民间疗法(2021年18期)2021-11-02
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
西南国防医药(2016年6期)2016-12-01
外语教学理论与实践(2016年1期)2016-06-11
癌症进展(2016年11期)2016-03-20
磁共振成像(2015年8期)2015-12-23
哈尔滨医药(2015年6期)2015-12-01
中国当代医药(2015年9期)2015-03-01
肿瘤预防与治疗(2014年4期)2014-11-24
