
O3/Fe2+均相催化体系及其效能强化综述
2021-08-17 03:06廖求文
净水技术 2021年8期
廖求文
(长沙矿冶研究院有限责任公司,湖南长沙 410012)
臭氧分子(O3)具有极强的氧化能力(2.07 eV)[1-2],被广泛应用于水与废水的脱色、消毒及难降解有机污染物的降解。根据氧化反应机理,O3氧化技术可分为直接氧化和间接氧化,其中,直接氧化即利用O3与有机物的氧化还原反应、环加成反应、亲电取代反应、亲核反应等,将大分子有机物分解为较小分子有机物。O3对芳香烃化合物、烯烃等含双键、三键等有机化合物具有选择性[2]。与间接氧化相比,直接氧化反应速率较慢[k=100~103mol/(L·s)],而O3进一步氧化降解小分子有机酸[如草酸(AO)、乙酸等]的反应速率更低,如O3与AO的反应速率常数为4×10-2mol/(L·s)[3],直接氧化难以实现完全矿化。
间接氧化是通过催化剂、紫外线照射、超声、-OH等方式促进O3分解产生·OH(2.8 eV),利用·OH的强氧化能力,快速、无选择的氧化降解有机物,并实现完全矿化[3],·OH氧化反应速率常数为108~1010mol/(L·s)。过渡金属离子,如Fe2+、Fe3+、Mn2+、Cu2+、Co2+、Ag+、Ni2+、Zn2+等被广泛研究用于催化O3氧化[4],由于Fe2+廉价易得、无二次污染、催化效率高,O3/Fe2+体系被深入广泛地研究。
本文对O3/Fe2+体系及其强化机理的研究现状进行综述,以期更深入地了解O3/Fe2+催化O3氧化体系及其强化,为O3/Fe2+体系及其强化的研究和应用提供借鉴。
1 O3/Fe2+催化氧化机理
Sauleda等[5]认为,O3/Fe2+遵循羟基氧化反应路径,提出了Fe2+催化O3分解产生·OH的机理,如式(1)~式(3)[5]。
Fe2++O3→ FeO2++O2
(1)
FeO2++H2O → Fe3++·OH+OH-
(2)
FeO2++Fe2++2H+→ 2Fe3++H2O
(3)
Fe2+与O3反应生成FeO2+,FeO2+与H2O发生水解反应产生·OH和副产物Fe3+、OH-;FeO2+与Fe2+反应生成Fe3+,阻断自由基链式反应;同时,Fe2+与·OH反应产生Fe3+。过量的Fe2+反而抑制O3催化氧化过程,如式(4)。
Fe2++·OH → Fe3++OH-
(4)
普遍认为,生成的Fe3+对O3氧化反应具有催化作用,而Fe3+对O3氧化催化反应机理则仍然存在争议。Beltrn等[6]认为,在pH值=2.5时,Fe3+与AO形成络合物,再经O3氧化去除,而非自由基氧化。竹湘锋等[7]认为,O3/Fe3+催化氧化AO的反应遵循自由基反应路径,Fe3+与AO形成草酸铁络合物[Fe3+(AO2-)n],在酸性条件下,Fe3+(AO2-)n能够促进O3分解产生H2O2,从而构成类Fenton体系,Fe3+(AO2-)n的氧化产物AO-被进一步氧化为CO2,如式(5)~式(6)。
AO-+O3+H+→ CO2+O2+·OH
(5)
·HO+AO-→ CO2+H2O
(6)
Pillai等[8]则认为,Fe3+促进O3分解产生·OH,如式(7)。
Fe3++O3→ FeO2++H++O2+·OH
(7)
Bader等[9]认为,在酸性条件下,O3分解生成H2O2,pH越低,H2O2生成量越大,金属离子催化剂存在时,形成类Fenton体系。
而王赫等[10]认为,Fe2+、Fe3+不具备催化O3降解AO的能力,γ-FeOOH催化O3产生的·OH在氧化降解AO过程中起到了主要作用,如式(8)~式(10)。
(8)
γ-FeO·+H2O → γ-FeOH+·OH
(9)
(10)
综上,O3/Fe2+/Fe3+体系催化O3氧化的能力及其反应机理仍然存在争议。
2 O3/Fe2+的影响因素
2.1 pH
2.1.1 pH影响氧化反应路径
单独O3氧化体系中,pH值<4时以直接氧化为主,pH值=4~9时直接氧化和间接氧化并重,pH值>9时以间接氧化为主[1-2,8]。Arslan等[11]研究表明,pH值=2.5、O3投加量[O3]=0.44 mg O3/(mg COD)时,O3吸收率达到峰值,且O3吸收速率随pH升高而显著提高。竹湘峰等[7]采用O3氧化AO,AO去除率随pH升高而提高,加入50 mmol/L叔丁醇(TBA)后,AO基本没有去除效果,说明在高pH条件下,O3氧化AO遵循自由基反应路径。
O3/Fe2+氧化体系中,pH、Fe浓度、氧化还原电位是影响Fe-H2O中Fe形态和组分的重要因素[12],从而影响催化O3氧化的有效成分和催化氧化反应机理。
2.1.2 pH影响催化O3氧化的有效成分
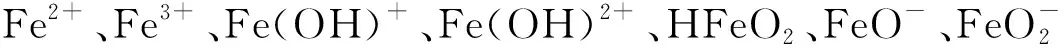
其中,Fe2+、Fe3+[7,11]和FeOOH(s)[10]可能是O3/Fe2+体系的主要有效成分,而pH影响Fe2+/Fe3+的水解平衡和Fe2+、Fe3+、FeOOH(s)等形态的浓度。根据Fe-H2O体系的优势区域图,当Fe浓度为0.1 mmol/L时,Fe3+主要存在于pH值=0~2.17中,且E=0.77~2 V。Fe2+主要存在于pH值=0~9.31中,且随着pH增加,Fe2+存在的E的范围变窄,当pH值=0~2.17时,Fe2+存在于E=-0.64~0.8 V中;当pH值=3.23时,Fe2+存在于E=-0.64~0.707 V中;当pH值=3.23~9.31时,Fe2+存在于E=-0.64~0.372 V中。根据Fe-H2O体系的Pourbaix图,当Fe浓度为0.1 mmol/L时,FeOOH(s)稳定分布在pH值=1.46~13.8中;而当Fe浓度为0.1×10-3mmol/L时,FeOOH(s)稳定分布在pH值=2.61~11.41中[12]。
Fe(OH)3的溶度积KSP[Fe(OH)3]=3×10-39,Fe(OH)2的溶度积KSP[Fe(OH)2]=8×10-16[14],Fe3+的溶解度极小,在较低pH条件下,Fe3+即可产生Fe(OH)3沉淀,pH值>3.7时达到“完全”沉淀。
在O3/Fe2+氧化体系高氧化电位环境中,Fe2+氧化为Fe3+,而Fe3+存在的pH范围窄,Fe(OH)3沉淀造成Fe2+和Fe3+的无效消耗,Fe2+和Fe3+均相催化效能降低。O3/Fe2+体系中FeOOH(s)的存在值得关注,但FeOOH(s)/O3体系最佳pH值=7[15],而O3/Fe2+体系最佳pH值在3左右。
在O3/Fe2+净化废水体系,Fe2+、Fe3+与有机物络合,Fe的形态更为复杂。竹湘峰等[7]认为,O3/Fe3+体系在pH值=5.5时对AO几乎没有去除效果,而在pH值=3时AO的去除率达到60%,是因为在酸性条件下,Fe3+与AO形成的Fe3+(AO2-)n催化O3进而氧化有机污染物。
杨鹏飞等[16]研究表明,O3/Fe2+体系氧化硝基苯的降解速率随着pH升高呈现先增大后变小的趋势,pH值≈2时反应速率较慢,以O3直接氧化为主;当pH值=3.5时,硝基苯的去除速率最大,达到93.2%;pH值>3.5时,硝基苯的氧化去除速率降低。Arslan等[11]研究表明,O3/Fe2+催化氧化合成染料废水的脱色和去除COD的研究中,pH值=3时,综合效果最佳;pH值=7.5时,CODCr去除率不超过10%。
2.2 Fe2+、Fe3+投加量
当pH一定时,Fe2+、Fe3+过量投加形成沉淀,造成Fe2+、Fe3+的无效消耗;同时,过量的Fe2+消耗·OH。Zhang等[13]研究表明,O3/Fe2+催化降解活性红2染料的速率常数K在Fe2+投加量[Fe2+]为0~3.6 mmol/L时,随着[Fe2+]变化而变化。K从[Fe2+]=0.0时的1 056 mol/(L·s)提高到[Fe2+]=0.9 mmol/L时的2 248 mol/(L·s);当[Fe2+]>0.9 mmol/L时,过量的Fe2+消耗·OH,导致活性红2降解效率降低,而当[Fe2+]大于pH值>4时的最佳投加量时,则产生Fe(OH)2、Fe(OH)3沉淀。竹湘峰等[7]研究表明,Fe3+投加量[Fe3+]过高或过低都不利于AO的降解,当[Fe3+]为25 mg/L、反应时间为30 min时,AO去除率>50%;当[Fe3+]为50 mg/L时,AO去除率反而下降。
2.3 O3投加量
O3/Fe2+氧化体系的O3传质属于伴随反应的气液传质过程,综合传质速率与气液物理吸附传质速率、Fe2+促进液相主体O3分解速率有关[1]。根据双膜传质理论,进入液相主体的O3浓度与气膜传质和液膜传质过程、气相主体O3浓度有关,当气相主体O3浓度较低时,随着[O3]增加,即进入液相主体的O3量增加,可提高O3/Fe2+体系催化氧化去除率。于忠臣等[17]研究了液相O3质量浓度和COD去除率与气相O3质量浓度的相关性,气相O3质量浓度由8 mg/L增加到14 mg/L时,液相O3质量浓度提高0.3 mg/L,CODCr去除率提高8%;气相O3质量浓度由14 mg/L增加到22 mg/L时,液相O3质量浓度提高1.9 mg/L,CODCr去除率提高18%。
当[O3]较大时,气膜传质和液膜传质过程成为O3/Fe2+体系的控制步骤,反应器形式影响气液传质物理吸附过程,强化气液传质、提高O3利用率。杨鹏飞等[16]利用超重力强化O3/Fe2+氧化降解硝基苯废水,在旋转填料床强化O3气液传质过程中,O3/Fe2+/RPB对硝基苯和COD的去除率,与O3-RPB相比分别提高了7.1%和27.51%,与O3/Fe2+/BR相比分别提高了27.2%和32.55%。
2.4 反应温度
根据阿伦尼乌斯定律,温度影响化学反应的活化能,从而影响反应速率常数K,O3直接氧化的活化能为35~50 kJ/mol,O3间接氧化的活化能为5~10 kJ/mol,当溶液O3浓度恒定时,反应稳定升高10 ℃,反应速率常数K增加1倍[1]。温度越高,O3溶解度越低,同时O3半衰期越短,加速O3的无效分解,不利于O3氧化。
Zhang等[13]研究表明,当反应温度从17 ℃上升到30 ℃时,O3催化氧化活性红2的反应速率常数K2从1 742 mol/(L·s)增加到2 818 mol/(L·s),活化能Ea=26.3 kJ/mol,频率因子Ko=9.32×107mol/(L·s)。温度提高13 ℃,K2仅增加到原来的1.6倍。
3 O3/Fe2+催化氧化及其效能强化
3.1 O3/Fe2+催化氧化效果
如表1所示,O3/Fe2+体系被广泛研究用于废水有机污染物的降解。杨鹏飞等[16]以初始浓度为175 mg/L的硝基苯模拟废水,分别利用O3和O3/Fe2+氧化降解试验,pH值=3.5、O3总投加量为2 g时,循环反应40 min后,硝基苯和CODCr去除率分别为92.40%和40.46%。而投加[Fe2+]=22.4 mg/L,硝基苯和CODCr去除率分别为99.50%和67.97%。

表1 O3/Fe2+应用及效果Tab.1 Application of O3/Fe2+and Effectiveness
Arslan等[11,18]分别利用O3和O3/Fe2+体系催化氧化净化染料废水,其中,用于净化CODCr初始质量浓度为3 790 mg/L的原水时,CODCr去除率分别为36%和41%;用于净化CODCr初始质量浓度为44 mg/L的生化处理出水时,去除率分别为11%和47.8%。
以上研究结果表明,在同等反应条件下,与单独O3氧化相比,O3/Fe2+体系显著提高有机污染物的降解效率。然而O3/Fe2+体系适用的pH范围狭窄,最佳pH值在3左右。反应存在最佳的[Fe2+]范围,过量Fe2+捕获·OH,效率降低,需要持续投加Fe2+,造成Fe2+消耗的同时产生铁泥。
在印度尼西亚,测绘地理信息主管部门是地理空间信息局(BIG)。BIG的主要职责是负责与地理空间信息相关的政务工作,不仅涉及国家大地控制网建设与基础地图生产等基础地理空间信息的生产和提供,还涉及专题地理空间信息的生产及协调工作。另外,BIG还与相关政府机构一起,负责建设印度尼西亚地理空间信息基础设施。印度尼西亚的地理空间信息基础设施涉及政策制定、制度建设、技术开发、标准制定和人才培养等方面的工作。
3.2 O3/Fe2+效能强化及效果
3.2.1 UV辅照
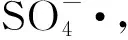
(11)
(12)
O3+H2O2→·OH+HO2·+O2
(13)
Sauleda等[5]利用UVA辐照强化O3/Fe2+降解苯胺和4-氯苯酚模拟废水,TOC去除率分别提高了27.5%和38%。于忠臣等[17]研究了不同催化O3体系(O3、UV/O3和Fe2+/UV/O3)对腈纶废水的降解特性,Fe2+/UV催化O3表现出极强的COD降解性能。Miklos等[20]利用UVA辐照强化O3/Fe2+降解2,4-二氯磷酸酯模拟废水,TOC去除率提高了27%。Skoumal等[21]利用UVA辐照强化O3/Fe2+降解废水中的扑热息痛,TOC去除率提高了33%,UV辐照强化O3/Fe2+降解有机废水的TOC去除率较高,显著提高O3/Fe2+体系的催化效果,pH值在3左右时,O3/Fe2+/UVA体系催化效果最佳,pH值=2和pH值=4时TOC去除率略低,pH值=6时TOC去除率更低。
3.2.2 H2O2
3.2.3 螯合剂
表2 O3/Fe2+及其强化效果对比Tab.2 Comparison of O3/Fe2+and the Enhancement Effect
3.3 羟胺强化O3/Fe2+体系的潜在应用
强化O3/Fe2+效能的关键在于抑制Fe(OH)2和Fe(OH)3沉淀、促进Fe3+还原Fe2+或利用多种机理协同降低Fe2+的无效消耗、提高拓宽适用的pH范围,达到强化效能的目的。在Fenton反应体系研究中[25-30],利用羟胺(HA)等还原剂促进Fe2+的再生,HA加速Fe3+/Fe2+转化,抑制Fe3+的积累和水解-沉淀[26-28],同时,HA对·OH的副作用较小,且不与H2O2反应[25],具有良好的效能强化效果。Gösta等[31]研究表明,HA与Fe3+发生氧化还原反应,生成Fe2+及含氮副产物,其含氮副产物种类与[Fe3+]∶[HA]有关,当Fe3+过量时,如[Fe3+]∶[HA]=2∶1时,含氮副产物以N2O为主;当[HA]过量时,即[Fe3+]∶ [HA]≤1∶1时,含氮副产物以N2为主。因此,通过调控[Fe3+]∶[HA]可防止引入含氮污染物。
HA/O3的研究表明,质子化羟胺(NH3OH+)与O3的反应速率为2 mol/(L·s),而非质子化羟胺(NH2OH)与O3反应的速率常数为(2.1±0.2)×104mol/(L·s)[29-30]。说明在HA/O3/Fe2+体系中,低pH条件下,质子化羟胺(NH3OH+)优先与Fe3+反应生成Fe2+。
以上分析表明,利用HA构建Fe3+/Fe2+循环,可能是实现O3/Fe2+效能强化的一种新方法。
4 结论与展望
(1)Fe2+易得、廉价、无毒、无二次污染、催化效率高,O3/Fe2+体系是一种具有运用潜力的O3高级氧化技术。然而O3/Fe2+/Fe3+催化氧化的有效成分和机理仍然存在争议。
(2)pH影响Fe2+、Fe3+在水溶液体系中的形态和水环境化学行为,是影响O3催化氧化机理及其效能的最主要的因素之一。Fe2+、Fe3+在水溶液体系中的形态和水环境化学行为,与投加量、pH、污染物浓度相关。
(3)Fe3+在UV的光解作用下,生成Fe2+和·OH,UV已被广泛运用于强化O3/Fe2+/Fe3+催化氧化体系。投加螯合剂,形成金属离子螯合物,多价金属离子(Mn+) 起到很好的隐蔽作用,降低金属离子在水相中的浓度与消耗速率,使金属离子实现持续的催化作用,从而提高催化效果。
(4)HA能加速Fe3+/Fe2+转化,在低pH条件下,NH3OH+催化 O3分解产生·OH,且HA对·OH的副作用较小,利用HA构建Fe3+/Fe2+循环,可能是实现O3/Fe2+强化的一种新方法。
猜你喜欢
工业安全与环保(2022年10期)2022-10-28
云南化工(2022年9期)2022-10-12
浙江大学学报(理学版)(2020年1期)2020-03-12
浙江大学学报(工学版)(2016年2期)2016-06-05
天津城建大学学报(2015年5期)2015-12-09
电源技术(2015年5期)2015-08-22
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
安徽农学通报(2015年2期)2015-02-12
应用化工(2014年1期)2014-08-16
