
不同发育时期大头金蝇蛹转录组测序分析
2021-08-20 03:36王启燕张红玲任峥刘玉波季晶焱黄江
法医学杂志 2021年3期
王启燕,张红玲,任峥,刘玉波,季晶焱,黄江
贵州医科大学法医学院 法医司法鉴定中心,贵州 贵阳550004
死亡时间推断是法医工作者首要解决的问题,利用案发现场昆虫推断死亡时间的主要依据是在一定环境条件下昆虫发育到某一特定阶段所需时间相对恒定,各种形态或生理特征变化有一定的规律性,根据该种昆虫所处的发育阶段可以推断其经历的发育时间,进而为死亡时间推断提供比较可靠的线索[1]。
转录组是指生物体的细胞或组织在一个特定状态下转录出来的所有RNA 的总和,包括信使RNA 和非编码RNA,转录组测序能够提供全部基因的表达信息和蛋白质的功能、相互作用的信息。昆虫转录组研究提供了一种快速、高通量、全面解读昆虫基因组信息的全新技术手段,为更全面、更深入地研究昆虫生命活动中相关基因功能、系统发生与进化、生长发育以及昆虫与其他生物相互作用等提供依据[2]。
大头金蝇是常见的嗜尸性蝇类,精确推断蛹的发育历期,对于推断死亡时间具有重要参考价值。为更好地了解大头金蝇蛹的转录组情况,挖掘更多与法医学相关的信息,本研究对收集到的6 个发育时期(6 d)的大头金蝇蛹进行转录组测序,通过差异基因的聚类模式分析、富集分析等方法,为深入分析大头金蝇蛹的发育调控、表观调控、环境适应以及法医学应用等问题奠定基础。
1 材料与方法
1.1 大头金蝇蛹的采集
以新鲜猪肺作诱饵,采用扣捕法采集成虫于夏季,经法医昆虫学专家参照《中国尸食性蝇类》[3]进行鉴定;饲养于50 cm×50 cm×50 cm 笼内,自然温度,以水、奶粉和白糖喂养。以新鲜猪肺为产卵介质,观察到成虫产卵后,将卵块取下,转移至饲养盆中用新鲜猪肺进行饲养,直至幼虫全部离食。从幼虫离食开始每1 h 观察1 次,化蛹开始收集,每24 h 收集1 次,每次收集3 粒蝇蛹,共收集6 d,直至开始羽化出成虫。蝇蛹来自同一只成虫,收集过程中平均温度为25.6 ℃。蝇蛹于-80 ℃保存备用。
1.2 RNA提取
对收集到的6 个发育时间(1~6 d)蝇蛹分别提取总RNA,均为3 次生物学重复,第1 天样本命名为CM1(CM1-1,CM1-2),第2 天的样本命名为CM2(CM2-1,CM2-2,CM2-3),以此类推。采用Trizol RNA 提取试剂(美国Invitrogen公司)分别提取18个样本的总RNA,利用NanoDrop 2000c分光光度计(美国Thermo Scien⁃tific 公司)对所提RNA 的浓度和纯度进行检测,琼脂糖凝胶电泳检测RNA 完整性,2100 型生物分析仪(美国Agilent 公司)测定RNA 完整值(RNA integrity num⁃ber,RIN)。RIN 值大于7.0 即可用于建库[4]。
1.3 转录组测序及数据组装
采用Illumina Hiseq4000 二代测序仪(美国Illu⁃mina 公司)进行测序,对获得的原始测序数据进行过滤,去除接头和含N(模糊碱基)比例超过10%的读长、碱基质量值小于20 的碱基修剪后,如剩余序列中仍有质量值小于10 的碱基则将整条序列剔出,舍弃去接头及质量修剪后长度小于20 bp 的序列,得到质量控制后的原始测序数据,为保证测序结果的准确度,分别统计质量值大于20、30 的碱基所占的百分比,即Q20、Q30 所占百分比。采用Trinity 软件(版本号trini⁃tyrnaseq_r20 140413;https://github.com/trinityrnaseq/tri nityrnaseq/wiki)进行序列组装,获得的转录本取最长的作为非重复序列基因(Unigene)序列。
1.4 功能注释
对转录本功能进行注释前,使用Trinity 软件提供的开放阅读框(open reading frame,ORF)预测流程(http://trinityrnaseq.sourceforge.net/analysis/extract_pro teins_from_trinity_transcripts.html)对组装得到的所有转录本序列进行基因预测。
将非重复序列基因使用美国国立生物技术信息中心(NCBI)的BLAST(Basic Local Alignment Search Tool,Version 2.2.25,期望值E<1×10-5)分别与非冗余蛋白库(Non-Redundant Protein Sequence Database,简称NR)、检索相互作用基因的搜索工具(Search Tool for the Retrieval of Interacting Genes,简称STRING;https://string-db.org)、SWISS-PROT 瑞士蛋白质数据库(包括Pfam,https://www.ncbi.nlm.nih.gov/search/all/?term=swissprot)、基因本体论(Gene Ontology,GO)数据库(http://www.geneon tology.org)、蛋白直系同源簇(Clusters of Orthologous Groups of proteins,简称COG)数据库(http://www.ncbi.nlm.nih.gov/COG)、京都基因和基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG;http://www.genome.jp/kegg/)进行比对获得相应的注释信息。
1.5 表达量分析与差异基因筛选
使用RSEM 软件(http://deweylab.biostat.wisc.edu/rsem/)对结果进行表达量统计,得到每个样品比对到每个基因上的序列数,对其进行每百万测序碱基中基因外显子每千个碱基长度中所包含的测序片断数(fragments per kilobase of exon model per million mapped reads,FPKM)转换,计算测序所得非重复序列基因在6 个发育时期大头金蝇蛹的表达水平,并以发育时期两两之间FPKM 表达量的比值的log2倍数绝对值大于1(即log2|FC|>1,其中FC 为fold change,表示两样品间表达量的比值)及错误发现率(false discovery rate,FDR)小于0.05 为标准进行差异基因筛选[5]。
1.6 差异基因表达模式聚类分析
将有显著差异的基因进行表达模式聚类分析,采用hcluster 聚类方法进行聚类分析,该方法用于聚类的算法是最长距离法(complete 算法),对收集到的15 个样本表达模式聚类分析时,不同样本间的比较采用Spearman 相关分析,差异基因表达模式聚类分析时,不同的差异基因间比较采用Pearson 相关分析。
1.7 差异基因的富集分析
按照差异基因参与的生物学过程、构成细胞的组分、实现的分子功能等进行分类,使用Goatools 软件(https://github.com/tanghaibao/GOatools)进行GO 富集分析,用KOBAS软件(http://kobas.cbi.pku.edu.cn/home.do)对KEGG 通路进行富集分析。富集分析均使用Fisher 精确检验,为控制计算的假阳性率,使用4 种多重检验方法(Bonferron 方法、Holm 方法、Šidák 方法和P值调整)对P值进行校正,当经过校正的P值≤0.05时,认为此GO 功能、KEGG 通路显著富集。
2 结果
2.1 测序与组装结果
提取18 粒蝇蛹的RNA,经2100 型生物分析仪检测,有15 个RNA 的RIN 值大于7.0,所提取的RNA 符合建库质量要求,其中第1 天、第3 天和第6 天各有1 粒蝇蛹的RNA 质量未达到建库质量要求。
通过Illumina Hiseq4000 二代测序仪进行转录组测序,对原始测序数据进行过滤,得到质控后原始测序数据用于后续分析,对质量修剪后的序列进行数据统计,Q30 碱基所占百分比为93.94%~96.03%(表1)。

表1 测序数据统计Tab.1 Sequencing data statistics
采用Trinity 软件对序列进行组装,对组装得到的转录组进行各项基础指标的统计(表2~3):共获得43 408 条Unigene,所有Unigene 的碱基个数为39 306 707 个,平均长度为905.52 bp;长度在1~400 bp的Unigene 数量最多,占总基因数量的42.95%,其次是601~800 bp,占13.07%。

表2 组装结果统计Tab.2 Summary of assembly results
2.2 功能注释
对拼接后获得的43408条Unigene,使用BLAST分别与NR、STRING、SWISS-PORT、Pfam、KEGG 数据库进行比对,其中通过NR、SWISS-PORT、Pfam、STRING、KEGG 数据库比对获得注释的结果分别有32 500、18 720、13 542、9 191、18 720 条Unigene,有4 450 个基因在所有数据库中均得到注释。
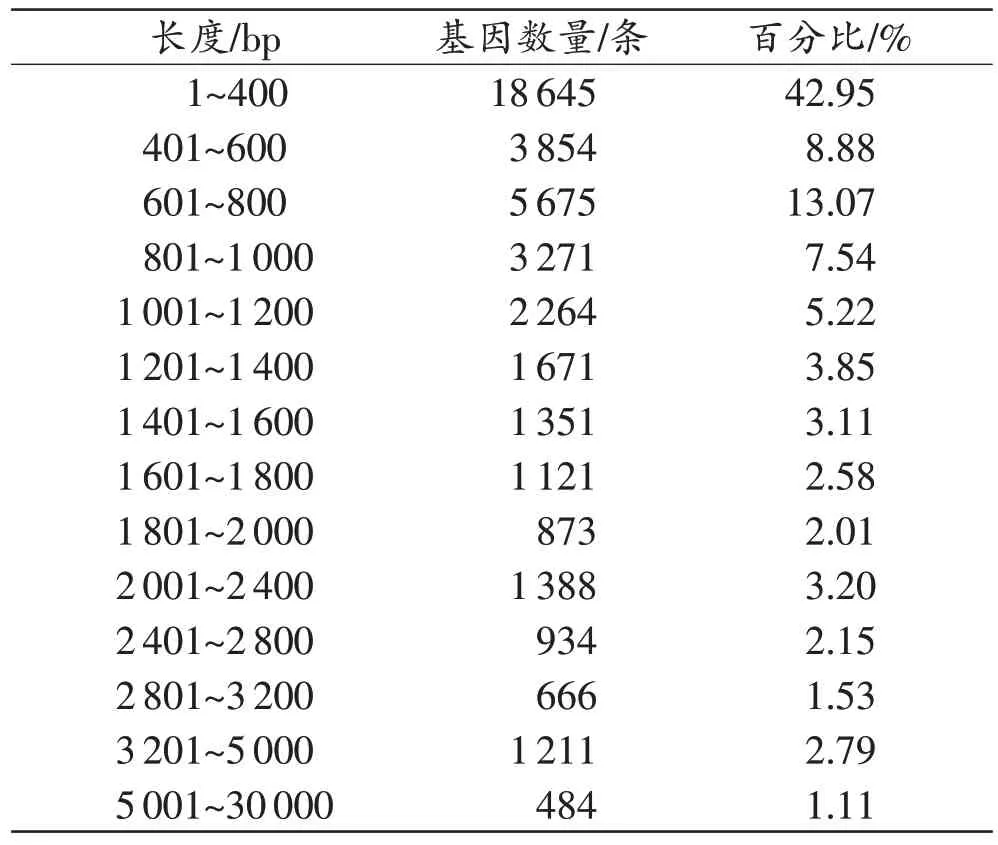
表3 组装Unigene长度分布Tab.3 Length distribution statistics of assembly Unigene
2.2.1 NR库注释结果
Unigene通过NR库相似序列匹配的近缘物种中,家蝇所占比例最高(9977条),其次为其他物种(8038条),之后依次为数据库中未收录物种(6 115 条)、地中海实蝇(1 350 条),占比最少的是甜橙(190 条),另外与黑腹果蝇、嗜凤梨果蝇、拟暗果蝇等物种均有相似序列,所占比例见图1。

图1 NR库比对物种分类图Fig.1 The species distribution compared with NR database
2.2.2 GO注释结果
利用GO 数据库对Unigene 的功能属性进行注释,包括生物学过程、细胞组分和分子功能3 大类,分别描述了基因产物可能参与的生物学过程、所处的细胞环境以及行使的分子功能。
共有15 979 条Unigene 得到注释。所有的匹配序列被进一步富集为52 个功能类别,其中生物学过程22 类、细胞组分17 类、分子功能13 类,在生物学过程中注释的基因数最多的是代谢过程有10 041 个,其次为细胞过程有9 649 个(图2A);细胞组分中注释基因数最多的是细胞和细胞部分,均有6 348 个(图2B);在分子功能中注释到基因数最多的是催化活性和结合活性,分别有8 166、8 021 个(图2C)。

图2 GO功能分类图Fig.2 GO functional classification
2.3 差异基因
不同发育时间两两进行比较,共获得差异基因45676个,其中第1天与第2天比较差异基因有3725个,第2 天与第3 天801 个,第3 天与第4 天1 153 个,第4天与第5 天1 040 个,第5 天与第6 天940 个,第2 天与第5 天差异基因数目最多5 307 个(表4)。

表4 不同发育阶段的差异基因数Tab.4 The number of discrepant genes in different growing periods
2.4 差异基因表达模式聚类分析
差异基因表达模式聚类图(图3)结果显示,第1天的2 个样本聚为一支,第2 天的3 个样本聚为一支,第3 天的2 个样本聚为一支,第6 天的2 个样本聚为一支,第4 天的3 个样本中有2 个聚为一支,另1 个与第5 天的3 个样本聚为一大支。
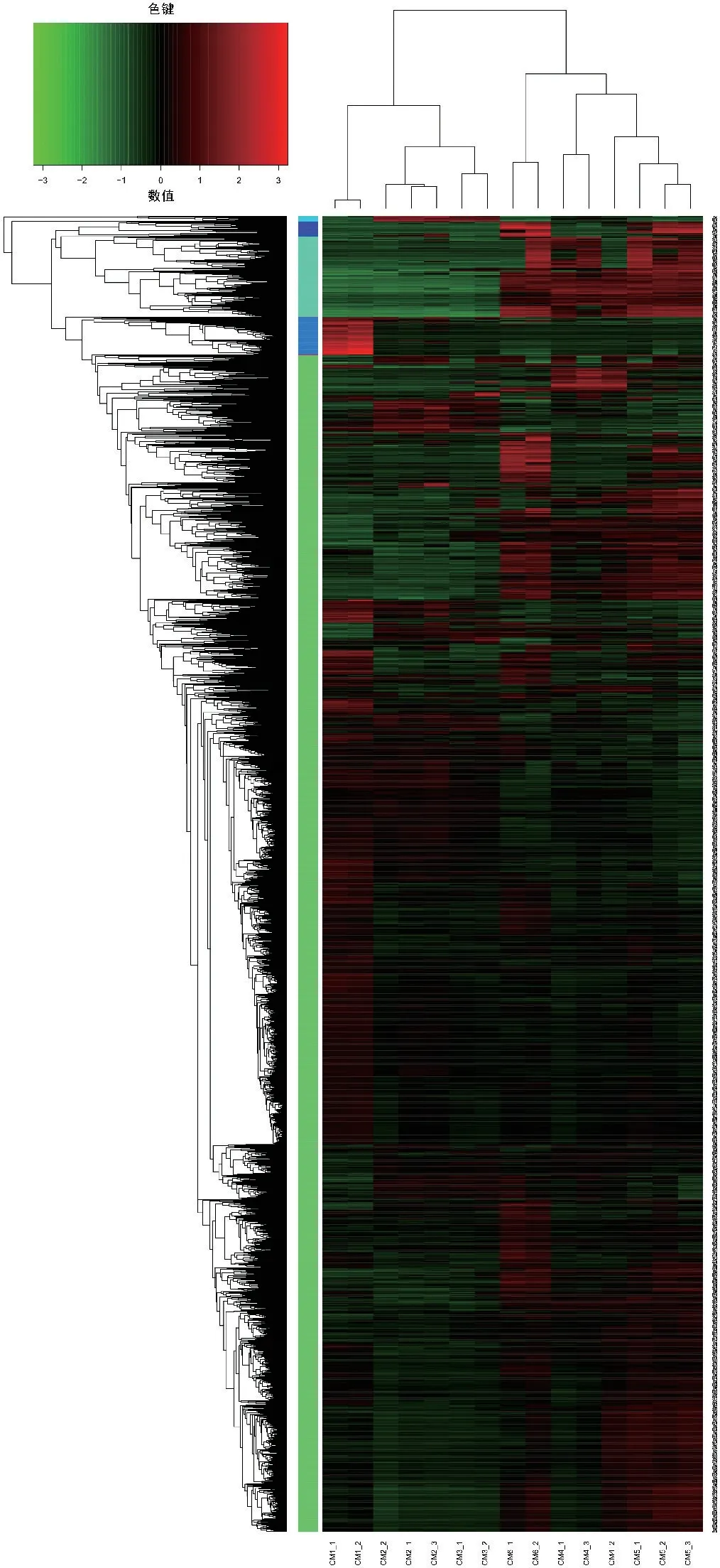
图3 差异基因表达模式聚类图Fig.3 Cluster of discrepant gene expression patterns
2.5 差异基因GO富集分析
第1 天与第2 天差异基因共富集了80 条GO 功能类别或细胞定位,生物学过程富集到43 条GO 功能或定位,其中富集的差异基因较多的是心脏收缩、自噬负调控、糖原分解代谢过程;细胞组分富集到16 条GO 功能或定位,其中富集到较多基因的是线粒体呼吸链复合体Ⅲ、核糖核蛋白复合体、易位子复合体;分子功能富集到21 条,其中富集到较多差异基因的是原肌球蛋白结合物、角质层的结构组成(图4)。

图4 差异基因GO富集柱状图Fig.4 Column of GO enrichment of discrepant genes
2.6 差异基因KEGG富集分析
通过分析得到大头金蝇蛹的差异表达基因KEGG富集情况,第1 天与第2 天相比,在角质、软木脂和蜡的生物合成,心肌收缩,叶酸合成,昆虫激素的生物合成,蛋白酶体等代谢通路富集最显著。后续富集变化情况见表5。

表5 差异基因KEGG通路富集情况比较Tab.5 Comparison of KEGG pathway enrichment of discrepant genes
3 讨论
常见的嗜尸性蝇蛹,从形态学上较难统一各发育阶段划分标准。GREENBERG[6]对伏蝇蛹的形态学研究结果表明,在发育过程中蛹壳内出现了11 个与发育时间相关的重要特征。王江峰等[7]在体视显微镜下,将蛹壳挑开后观察蛹内虫体的形态学变化,将大头金蝇蛹划分为7 个阶段,同时对在不同温度条件下达到发育阶段所需的时间进行了研究。将亮绿蝇蛹形态学随发育时间变化划分为12 个发育阶段[8]。国外有学者[9]将红头丽蝇的蛹期划分为15 个发育阶段(从化蛹开始,每24 h 收集1 次,每次收集5 个样本,直至羽化出成虫),进行从头测序,通过生物信息学分析筛选出各发育阶段具有差异性表达的基因并进行比对,该研究结果证实了蛹在不同时期的基因表达有显著差异,因此,有望通过该方法对不同发育时期的蝇蛹进行鉴别。周昊等[10]对棕尾别麻蝇蛹3 个不同发育时期进行转录组测序,并从中筛选出酪氨酸羟化酶、CBM-14基因进行实时荧光定量PCR,结果显示差异基因分析的表达模式一致。有学者[11]通过实时荧光定量PCR(real-time PCR)研究不同发育时期不同温度条件下大头金蝇蛹内蜕皮激素受体基因ecr、白眼基因white及热休克蛋白基因hsp70的基因表达情况,结果显示,这些基因具有规律性变化,且表达水平与温度有关,可作为大头金蝇蛹期推断的分子指标。有研究结果[12]显示,Hsp23、Hsp24和1_16基因的mRNA水平在巨尾阿丽蝇蛹期呈现出特定的时间变化规律,可作为巨尾阿丽蝇蛹期推断的分子指标。另外,有研究[13]结果显示,Hsp90、A-alpha、AFP、AFBP可作为分子指标用于更准确地估计棕尾别麻蝇蛹的发育年龄。WANG 等[14]对大头金蝇的卵、幼虫及成虫均进行了转录组测序,并筛选出与嗅觉相关的嗅觉蛋白进行分析。
为深入分析大头金蝇蛹的发育调控、表观调控、环境适应等问题,以便较好地为法医学应用奠定基础,本研究在自然环境中(平均温度为25.6 ℃)收集不同发育时期(历时6 d)大头金蝇蛹进行转录组测序,与杨玉璞等[15](26 ℃历时5.57 d)、马玉堃等[16](24 ℃历时6 d)的研究结果一致。
本研究通过对不同发育时期大头金蝇蛹的转录组测序分析,共获得43 408 条Unigene,获得注释最多的是NR库(32500条),STRING库注释最少(9191条),将不同发育时期蛹两两比较,差异表达基因数从801~5 307,差异表达基因总数45 676 个。从表达模式聚类图可看出,同一天的样本距离较近,均能较好地聚为一支,表明每天的样本中所有基因的表达模式接近,即基因的表达量变化趋势接近。富集分析方法通常是分析一组基因在某个功能节点上是否出现过,可提高研究的可靠性,能够识别出与生物现象最相关的生物学过程,本研究对转录组数据进行GO 富集分析,结果表明:第1 天与第2 天差异基因在角质层的结构组成、心脏收缩、核糖核蛋白复合体等显著富集。在KEGG 通路富集结果中角质、软木脂和蜡生物合成通路,心肌收缩,叶酸合成,昆虫激素的生物合成,蛋白酶体等显著富集。差异基因的GO 富集与KEGG 富集结果一致,第1 天与第2 天相比,均集中在角质层生物合成、心脏收缩、蛋白质合成及昆虫激素的生物合成等过程,其中角质、软木脂和蜡的生物合成代谢通路在后续第2~5 天的KEGG 通路富集分析均显著富集,说明角质、软木脂和蜡的生物合成、几丁质代谢以及昆虫的激素合成等与大头金蝇蛹的生长发育和变态过程密切相关,这与其他研究[17-19]结果均一致。
基于以上转录组信息,后期可通过对各个时期显著富集的差异基因进行分析,筛选出与大头金蝇蛹发育时间相关性最好的分子标记,结合形态学变化特点,对其结构功能、生物学意义及法医学应用价值等进行研究,通过差异表达基因在不同发育时期的表达模式,以及参与的具体代谢通路等进行深入分析,探索大头金蝇蛹发育机制,分析候选分子标记与发育时间的相关性,为蝇蛹的发育阶段的划分提供理论依据,为案发现场的蝇蛹推测死亡时间提供科学依据。此外,大头金蝇分布广泛,不同地理环境下的基因组水平存在差异,根据本研究获得的转录组数据,通过分析其SNP、STR 等信息,与其他地区大头金蝇的序列进行比较,获得贵阳地区大头金蝇特有的SNP、STR位点,为有死后移尸嫌疑的案件提供证据,同时与其他常见嗜尸性蝇类比较,可获得大头金蝇特有的遗传信息,为进行嗜尸性蝇类的种属鉴定提供参考。
猜你喜欢
中国人兽共患病学报(2022年9期)2022-10-19
科学导报(2021年29期)2021-06-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国中药杂志(2017年15期)2017-08-30
中国中药杂志(2017年15期)2017-08-30
江苏农业科学(2017年5期)2017-04-15
科学中国人(2016年17期)2016-01-28
湖北农业科学(2014年3期)2014-07-21
