
高效液相色谱法测定口服液体制剂中12种添加剂的含量
2021-09-01 03:13辜慧李道霞何成军杜钢四川省食品检验研究院成都611731
中南药学 2021年7期
辜慧,李道霞,何成军,杜钢(四川省食品检验研究院,成都 611731)
口服液体制剂易被微生物污染而发霉变质,为保障该类制剂临床用药安全,需添加适量的防腐剂,抑制微生物的生长繁殖,达到有效的防腐目的。另外,还需添加适量甜味剂及色素,改善制剂口感及外观,提高患者依从性[1]。然而,添加剂的超范围使用、过量添加会对人体造成一定的伤害。人工合成色素、甜味剂和防腐剂在市场上已有滥用的趋势,给药品安全带来了隐患,对消费者的健康造成了直接的威胁。目前,国内口服液体制剂生产企业众多,所加添加剂的种类与浓度亦各不相同,关于添加剂的测定方法文献均有报道[2-7],未见有同时测定人工合成色素、甜味剂和防腐剂的方法报道。为了提高检验效率,节约成本,更全面地控制口服液体制剂的安全性,本研究建立了一种同时测定口服液体制剂中12种添加剂的高效液相色谱(HPLC)法。
1 仪器与试药
LC-20AT 型高效液相色谱仪,配二极管阵列检测器(日本SHIMADZU 公司);ME204 分析天平(十万分之一,瑞士METTLER TOLEDO 公司);FB15065 超声波发生器(美国Fisher 公司);Milli-Q 型高纯水发生器(美国Millipore 公司);苯甲酸(批号:B0001649,浓度:1000 μg·mL-1)、山梨酸(批号:B0002741,浓度:1000 μg·mL-1)、酸性红(批号:C0005676,纯度:90.7%)、脱氢乙酸(批号:B0003452,纯度:99.9%)、糖精钠(批号:B0001955,浓度:1000 μg·mL-1)(BePure);柠檬黄、日落黄、苋菜红、胭脂红、新红、诱惑红及亮蓝(A CHEMTEK INC,批号:S027451,浓度:1000 μg·mL-1);甲醇、乙酸铵(色谱纯,美国Fisher 公司);水为超纯水。
2 方法与结果
2.1 色谱条件
色谱柱:Waters XBridge C18(250 mm×4.6 mm,5 μm);流速:0.8 mL·min-1;柱温:35℃;检测波长:230 nm;进样体积:10 μL;流动相A 为20 mmol·L-1乙酸铵溶液,流动相B 为甲醇,梯度洗脱程序见表1。
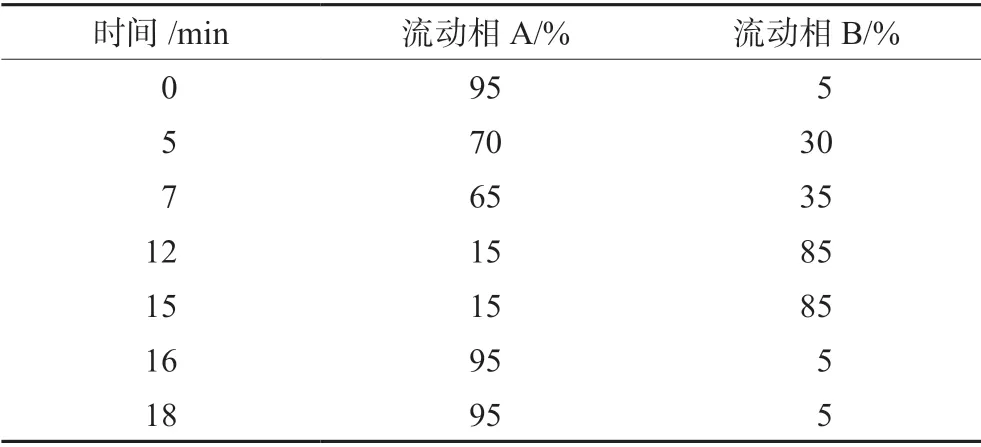
表1 梯度洗脱程序表Tab 1 Gradient elution program
2.2 溶液的配制
2.2.1 混合对照品溶液 分别精密称取酸性红、脱氢乙酸对照品约10 mg,分别置10 mL 量瓶中,用水超声溶解并定容至刻度,摇匀,分别作为酸性红对照品储备液、脱氢乙酸对照品储备液。其余组份对照品储备液均从对照品公司购得。再分别精密量取各对照品储备液1 mL,置同一50 mL 量瓶中,加水稀释至刻度,摇匀,作为混合对照品溶液。
2.2.2 供试品溶液 以布洛芬混悬液为例,精密量取样品1 mL,置10 mL 量瓶中,加水适量,超声处理10 min,再加水定容至刻度,摇匀,经0.45 μm 滤膜过滤,取续滤液作为供试品溶液。
2.2.3 阴性样品溶液 取阴性样品(蛋白酶合剂)按“2.2.2”项下方法制备阴性样品溶液,备用。
2.3 方法学考察
2.3.1 专属性考察 分别量取“2.2”项下的空白溶剂(水)、混合对照品溶液、阴性样品溶液、供试品溶液,按“2.1”项下色谱条件进样10 µL,记录色谱图,见图1。结果色谱图基线稳定,峰形对称,分离度较好,各峰保留时间适中,能够满足检测需要,说明本试验的专属性良好。

图1 典型HPLC 色谱图Fig 1 Typical chromatogram by HPLC
2.3.2 线性关系考察 分别精密量取不同体积的各对照品储备液,加水稀释成质量浓度分别为0.5、1、2、5、10、20 μg·mL-1的混合对照品溶液,精密吸取10 μL 注入液相色谱仪,以各组分的峰面积Y为纵坐标,进样质量浓度X(μg·mL-1)为横坐标,绘制标准曲线,结果见表2。
2.3.3 检测限及定量限考察 取混合对照品溶液,逐级稀释成系列质量浓度,分别进样10 μL,当峰高为基线噪音的3 和10 倍量时,测得各添加剂的检测限及定量限,结果见表2。

表2 12 种添加剂的回归方程、相关系数、线性范围、检测限与定量限Tab 2 Regression equation,correlation coefficient,linearity,LOD and LOQ for 12 additives
2.3.4 精密度考察 取混合对照品溶液,按“2.1”项下色谱条件下连续进样6 次,苯甲酸等12 种添加剂峰面积的RSD为0.11%~0.85%(n=6),表明仪器精密度良好。
2.3.5 重复性考察 取一批样品(检出柠檬黄、苋菜红、胭脂红),按“2.2.2”项下方法前处理,平行制备6 份,按“2.1”项下色谱条件检测,计算得样品中柠檬黄平均含量为0.52 μg·mL-1,苋菜红平均含量为3.11 μg·mL-1,胭脂红平均含量为5.88 μg·mL-1,RSD分别为1.8%、1.4%、0.99%(n=6),表明方法重复性良好。
2.3.6 回收试验 以未检出添加剂的阴性样品为基质,按低、中、高浓度分别精密加入混合对照品溶液(质量浓度为20 μg·mL-1)0.5、1.0、5.0 mL,按“2.2.2”项下方法处理,制备约相当于1、2、5 μg·mL-1的回收试验溶液,每个浓度平行制备6 份。按“2.1”项下色谱条件下进行检测,并计算回收率及RSD,结果见表3。

表3 12 种添加剂的平均回收率(n=6)Tab 3 Average recovery of 12 additives (n=6)
2.3.7 稳定性试验 取回收试验中的低、中、高浓度的溶液,室温放置,分别于0、6、12、24、48 h 进样,各组分峰面积的RSD均小于2.0%,表明12 种添加剂在48 h 内均稳定。
2.3.8 样品测定 取10 批抽检的口服液体制剂,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件测定,结果见表4。

表4 口服液体制剂中检测出的各成分含量测定结果(μg·mL-1,n=3)Tab 4 Content determination of various constituent in oral liquid preparations (μg·mL-1,n=3)
3 讨论
3.1 流动相的选择
液相色谱方法测定目标化合物时,多在流动相中加入缓冲盐,试验通过优化流动相梯度洗脱程序获得合适的保留时间、峰形以及分离效果。本试验参考文献[8-10],流动相选择乙酸铵溶液-甲醇系统,考察了流动相中乙酸铵的质量浓度分别为10、20、50 mmol·L-1时对12 种添加剂色谱行为的影响。结果表明乙酸铵质量浓度为20 mmol·L-1时,12 种添加剂的色谱峰峰形尖锐,分离度较好。故最终选择流动相为20 mmol·L-1乙酸铵溶液-甲醇体系,该流动相体系也可在检出添加剂后,直接用于LC-MS/MS 作进一步分析确认。
3.2 检测波长的确认
利用二极管阵列检测器对苯甲酸、山梨酸、糖精钠、脱氢乙酸、柠檬黄、日落黄、苋菜红、胭脂红、新红、酸性红、诱惑红及亮蓝分别进行波长扫描。其中苯甲酸、山梨酸、糖精钠、脱氢乙酸最大吸收波长均在紫外光区,柠檬黄、日落黄、苋菜红、胭脂红、新红、酸性红、诱惑红及亮蓝虽然最大吸收波长均在可见光区,但紫外光区也有较强吸收。综合分析12 个标准物质的吸收曲线,各组分在 220~240 nm 均有较强吸收,故选择230 nm 作为检测波长,在此波长下基线平稳、噪声小,各组分均具有较高灵敏度。
3.3 色谱柱的选择
本文参考文献[10-12],考察了色谱柱Waters XBridge C18(250 mm×4.6 mm,5 μm)、Agilent ZORBAX SB-C18(4.6 mm×250 mm,5 µm)、Eclipse XDB-C18(4.6 mm×250 mm,5 μm)对目标化合物分离度及峰形的影响,最终选择 Waters XBridge C18色谱柱,结果12 个目标化合物可以达到分离测定的要求,且准确可靠、重复性好。
3.4 其他
本试验采用HPLC 法同时测定口服液体制剂中的苯甲酸、山梨酸、糖精钠、脱氢乙酸、柠檬黄、日落黄、苋菜红、胭脂红、新红、酸性红、诱惑红及亮蓝的含量,该方法操作简便、精密度好、回收率高,能满足检测要求,适用于口服液体制剂中人工合成色素、甜味剂和防腐剂的检查。结果发现目前国内一些药品中仍存在着添加剂滥用的现象,存在较大的安全隐患,需要进行严格监管。
猜你喜欢
绥化学院学报(2022年8期)2022-08-26
食品安全导刊(2020年21期)2020-09-07
农家科技中旬版(2019年9期)2019-10-08
山西农业科学(2019年6期)2019-06-19
小小说月刊(2019年4期)2019-04-18
山东化工(2019年2期)2019-02-21
山东工业技术(2016年13期)2016-06-29
母子健康(2015年7期)2015-09-24
火花(2015年1期)2015-02-27
理科考试研究·高中(2014年9期)2014-09-22
