
超高效液相色谱-四极杆/静电场轨道阱高分辨质谱法快速筛查及测定蜂蜜中20 种植物源毒性成分
2021-09-14 00:52刘珈玲
食品工业科技 2021年17期
韦 环,刘珈玲,廖 强
(广西-东盟食品检验检测中心,广西南宁 530021)
我国《蜜蜂产品术语》(GB/T 20573-2006)对蜂蜜的定义为:是蜜蜂采集植物的花蜜,蜜露等分泌物,与自身分泌物结合后在巢脾内经过充分酿造而成的天然甜物质。不仅口感香甜,且具有润肺止咳、润肠通便、清热解毒等功效[1−2],因此深受人们的喜爱。蜂蜜本无毒,但在生产过程中被污染或蜜蜂采集有毒花蜜酿制而成的蜂蜜可能会引起中毒。尤其野生蜂蜜是食物中毒事件的主要致病因子之一[3−4]。建国以来广西、云南、贵州、福建多地都相继报道了食用野生蜂蜜中毒事件[5−7]。食用有毒蜂蜜后会引发一系列的中毒症状,如恶心、呕吐、腹泻、四肢麻木、血压下降、呼吸中枢麻痹等,严重将引发休克甚至死亡[4,6,8],且中毒程度及症状与蜂蜜所含的毒素种类和含量密切相关。有毒蜂蜜中所含毒素主要来源于蜜蜂采集了有毒源性的植物花粉,如断肠草、雷公藤和狼毒等[6,8−10];雷公藤、断肠草、洋金花、乌头、千里光等所含毒性物质主要为生物碱和萜类化合物,如乌头中的二萜类生物碱:乌头碱[11];博落回中的异喹啉类生物碱:小檗碱、原阿片碱和别隐品碱[12];菊科(如千里光属和泽兰属)中的吡咯里西啶类生物碱:千里光宁碱、千里光菲林生物碱、倒千里光碱[10,13];胡蔓藤属中的吲哚类生物碱:钩吻碱、钩吻素已、胡蔓藤碱丙、胡蔓藤碱丁[14];马桑属中的萜类化合物马桑亭[15]等。
《食品安全国家标准 蜂蜜》(GB 14963-2011)中明确规定“蜜蜂采集植物的花蜜、分泌物或蜜露应安全无毒,不得来源于雷公藤、博落回、狼毒等有毒蜜源植物。”但并未列出具体植物源毒性成分和相关检测方法。目前国内对植物中相关毒性物质的检测方法主要有液相色谱法[16−17]、气相色谱法[18]、酶联免疫吸附分析[19−20]、薄层层析测定法[21−22]、气相色谱-质谱法[23−25]、液相色谱-质谱法[15−16,26−28]等。液相色谱法检测灵敏度较低,适合于含量较高的样品如中草药;酶联免疫吸附分析法酶活性易损失、无法对各类成分进行准确定量分析;气相色谱-质谱法及液相色谱-质谱法均可实现微量衡量同时多组分分析,是目前司法鉴定主要方法,但气相色谱-质谱法不适用于热不稳性及高沸点化合物,液相色谱-质谱法可有效解决这样难题,但传统的三重四极杆质谱需要标准物质实现定性确证。高分辨质谱仪可依据其高质量准确度、高质量分辨率的全扫描数据在不需要标准物质的情况下实现定性确证,在食品定向和非定向筛查中应用广泛。
目前我国关于蜂蜜的质量安全研究多以兽药残留[29]、农药残留[30]、重金属[31]和真伪鉴别[32−33]为主,对于蜂蜜中植物源性毒性成分的研究甚少,仅有少数对有毒蜜源性植物雷公藤、昆明山海棠和博落回中毒性成分及其分析方法进行了报道[6,10,34−35],但是研究成分单一,各方法相对独立且选择性差、定性能力弱。食物中毒作为突发公共卫生事件,往往需要在短时间内查明中毒原因,对于行踪不定的野生蜜蜂其蜜源难以通过其行踪来推断其蜜源植物。因此,迫切需要建立蜂蜜中植物源性多种毒性物质的快速筛查方法。四极杆/静电场轨道阱高分辨率质谱仪(QExactive)具有良好的定性分析和高分辨率的特点,在单个分析周期内即可完成对样品高通量、高精度的一级、二级扫描,为化合物的准确鉴定提供了客观依据。
本研究以固相萃取法纯化样品,应用高效液相色谱-四极杆/静电场轨道阱高分辨率质谱联用技术,建立蜂蜜中20 种植物源毒性成分的快速筛查和测定方法,基于高分辨质谱的精准分子量和多级碎片信息确立了20 种植物源毒性成分的质谱数据库。该方法快速、准确、通量高,为进一步开展蜂蜜溯源,安全性评价和监测评估食用蜂蜜中毒提供参考,保障了蜂蜜的食用安全和提升了质量控制水平。
1 材料与方法
1.1 材料与仪器
标准物质野百合碱(CAS:315-22-0,纯度99.48%)、钩吻碱(CAS:509-15-9,纯度98.19%)、东莨菪碱氢溴酸盐(CAS:114-49-8,纯度98.7%)、倒千里光碱(CAS:480-54-6,纯度98.02%)、钩吻素子(CAS:1358-76-5,纯度98.34%)、千里光宁(CAS:130-01-8,纯度99.48%)、钩吻碱己(CAS:82354-38-9,纯度99.53%)、马桑亭(CAS:91653-75-7,纯度98.96%)、闹羊花毒素II(CAS: 26116-89-2,纯度99.79%)、闹羊花毒素V(CAS:37720-86-8,纯度99.82%)、原阿片碱(CAS:130-86-9,纯度99.56%)、A-别隐品碱(CAS:485-91-6,纯度98.58%)、次乌头碱(CAS:6900-87-4,纯度99.04%)、乌头碱(CAS:302-27-2,纯度98.52%) 美国斯坦福化学公司;春千里光碱(CAS:72755-25-0,纯度99.72%)、春千里光碱N 氧化物(CAS:101687-28-9,纯度99.87%)、N-氧化芝麻菜叶千里光(CAS:123864-94-8,纯度99.35%)、胡蔓藤碱乙(CAS:82375-29-9,纯度98%)
德国PhytoLab 公司;千里光菲灵碱(CAS:480-81-9,纯度99.53%) 成都德思特生物技术有限公司;闹羊花毒素III(CAS:26342-66-5,纯度98.6%) 上海源业生物科技有限公司);SPE 小柱:混合型阳离子交换固相萃取小柱(MCX,60 mg,3 cc) 美国沃特斯有限公司;甲醇、乙腈、甲酸 均为色谱纯,德国默克公司;其余 均为分析纯;实验用样品为蜜博士百花蜂蜜(批号20190115) 广西蜜博士蜂业有限责任公司生产,经预检验样品中不含有上述20 种化合物),其余66 批均购于广西南宁、崇左、贵港、来宾、钦州等地农贸市场或蜜蜂养植基地。
Ultimate 3000 超高效液相色谱仪-Q-Exactive四极杆/静电场轨道阱高分辨质谱仪 美国Thermo Fisher Scientific 公司;XS205 DU 电子分析天平 瑞士梅特勒-托利多公司;Multi Reax 全自动振荡仪德国Heidolph 公司。
1.2 实验方法
1.2.1 标准溶液的配制 准确称取野百合碱、钩吻碱、东莨菪碱氢溴酸盐、倒千里光碱、钩吻素子、千里光宁、钩吻碱己、马桑亭、闹羊花毒素II、闹羊花毒素III、闹羊花毒素V、原阿片碱、别隐品碱、次乌头碱、乌头碱、春千里光碱、春千里光碱N 氧化物、N-氧化芝麻菜叶千里光、胡蔓藤碱乙、千里光菲灵碱等20 种标准物质各约10 mg(精确至0.1 mg),分别置于10 mL 容量瓶中,用甲醇溶解并定容至刻度,配制成约1.0 mg/mL 的标准储备溶液。分别精密量取各标准储备溶液1 mL,置于同一100 mL 容量瓶中,用甲醇稀释并定容至刻度,得到10 μg/mL 混合标准中间溶液。
1.2.2 样品处理 称取1 g(精确至0.01 g)样品,置于15 mL 聚丙烯离心管中,加入水5 mL,振荡使溶解,将所有溶液通过混合型阳离子交换固相萃取小柱使用前依次用5 mL 甲醇、5 mL 水活化)中,弃去洗脱液,然后用10 mL 水淋洗小柱,弃去,再用9 mL 5%氨化甲醇进行洗脱,收集洗脱液至10 mL 容量瓶中,使用5%氨化甲醇定容,混匀后经0.22 μm 有机相滤膜过滤,即得。
1.2.3 色谱条件
1.2.3.1 液相条件 色谱柱:Thermo GOLD AQC18柱(2.1 mm×100 mm,1.9 μm);柱温:30 ℃;进样体积:2 μL;流速:0.3 mL/min;流动相:A 为含0.1%甲酸的10 mmol/L 甲酸铵溶液,B 为0.1%甲酸乙腈。梯度洗脱程序:0.0~5.0 min,5%~20% B;5.0~10.0 min,20%~45% B;10.0~13.0 min,45%~90% B;13.0~14.0 min,90 % B;14.0~15.0 min,90%~5% B;15.0~20.0 min,5% B。
1.2.3.2 质谱条件 离子源采用HESI 源(heated ESI),喷雾电压为3.5 kV(+)/3.0 kV(−),透镜电压为50 V,离子传输管温度:320 °C,鞘气流量:35 arb;辅助气体流量:10 arb;辅助气温度:300 °C。扫描方式:采用正、负离子同时扫描,采集模式:Full MS/dd-MS2模式,其中一级全扫描的分辨率:70000 FWHM,扫描范围:m/z 50~1200,自动增益(AGC):3×106,最大驻留时间:100 ms;二级扫描分辨率:17500 FWHM,自动增益(AGC):2×105,最大驻留时间: 50 ms;质荷比窗口宽度(Isolation Window):m/z 2.0;顶点激发(Apex tigger):4~8 s;排除同位素峰(Exclude isotopes)设为“on”,动态排除(Dynamic Exclusion)设为6.0 s,归一化碰撞能量(NCE)为20%、40%、60%。20 种物质其他条件详见表1。
1.3 数据库的建立
精取“1.2.1”标准使用液1.0 mL,置于100 mL 容量瓶中用甲醇定容至刻度;按“1.2.3”条件进样,将得到的高分辨质谱数据通过Xcalibar 软件得到目标物的准确质量数、保留时间、二级碎片离子等信息,将信息输入到筛查软件TraceFinder 下,建立质谱数据库(Compound Database)。
1.4 定量检测
本实验采用Full MS/dd-MS2 模式,选择母离子(见表1)进行外标法定量。

表1 20 种植物源性毒性成分的分子式、保留时间、精确质量数、质量数偏差及主要二级离子Table 1 Molecular formula, retention time, precise mass number, mass number deviation and main secondary ion mass number of 20 plant-derived toxic
2 结果与分析
2.1 色谱条件的优化
由于本次实验20 种化合物来源于多种植物,且大部分为生物碱类成分,极性差异较大。故本实验分别考察了Thermo GOLD AQ-C18、Agilent HILIC(150 mm×2.1 mm,3 μm)、ACE Excel2 C18-PFP(150 mm×2.1 mm,3 μm)三种类型的色谱柱,结果Thermo GOLD AQ-C18在20 种化合物中分离效果和峰形最佳;在流动相的考察中,结果发现乙腈离子化能力强于甲醇,且基线噪音较低;0.1%甲酸乙腈能有效改善峰形及离子化效果,故选0.1%甲酸乙腈作为有机相;水相中加入0.1%甲酸后离子化效率、化合物响应值更高;加入10 mmol/L 甲酸铵后能兼顾负离子扫描时也得到较好的响应,故最终0.1%甲酸(含10 mmol/L 甲酸铵)作为水相流动相。
2.2 质谱条件优化
2.2.1 质谱参数的优化 本实验使用了Full MS/dd-MS2采集模式,该模式首先进行一级全扫描,然后对指定的前级离子做进一步的二级扫描。在实验室中,对一级全扫描的质谱参数先后考察了35000、70000及140000 的分辨率对质谱信号的影响,结果显示使用140000 的分辨率一级离子的灵敏度会显著降低,这可能是由于过高的分辨率会显著降低扫描速度,造成一级扫描点数不足,造成色谱峰形变差,影响分析,故选择了一级质谱扫描采用70000 的分辨率。
2.2.2 物质电离方式的优化 在1.2.3 质谱条件下,闹羊花毒素II、闹羊花毒素III 易发生离子源内裂解,闹羊花毒素II m/z=410.22990 考虑到其结构可能在离子源内裂解失去-C2H3O2, 形成了稳定的双键得到m/z=297.18439(见图1);闹羊花毒素III m/z=329.19587 在离子源内裂解失去4 个H2O 后形成了稳定的双键,得到m/z=297.18439(见图2)。闹羊花毒素V[M+H]+电离模式下没有加氢峰,而在[M+Na]+电离模式下有极强的加钠峰;马桑亭在[M-H]−电离模式下能得到很好的减氢峰,其它17 种植物源性毒性成分在[M+H]+电离模式下均能得到很好的加氢峰。
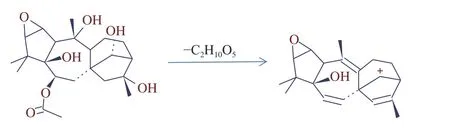
图1 闹羊花毒素II 的裂解图Fig.1 Chinese azalea flowers toxins II cracking figure
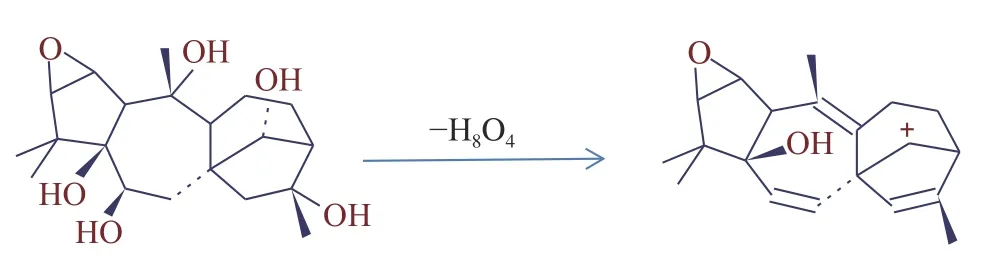
图2 闹羊花毒素IIII 的裂解图Fig.2 Chinese azalea flowers toxins IIII cracking figure
2.3 固相萃取小柱的选择
由于20 种植物源性毒性成分多数生物碱等碱性物质,故本实验选择了混合型阳离子交换固相萃取小柱(MCX)、混合型弱阳离子交换固相萃取小柱(PCX)、Waters Oasis HLB 三种类型对生物碱有效好保留的固相萃取小柱进行前处理考察。结果发现混合型阳离子交换固相萃取小柱(MCX)对20 种植物源性毒性成分具有高的选择性和灵敏度,均获得了很好的净化效果;而Waters Oasis HLB 对千里光菲灵碱的保留较差,回收率只有62.81%;从图3 中能看出 PCX 对钩吻碱、千里光菲灵碱、钩吻素子、千里光宁(千里光碱)、春千里光碱、钩吻碱己、原阿片碱、胡蔓藤碱乙、别隐品碱、次乌头碱、乌头碱保留较差,回收率都无法满足实验要求;故首选了Waters Oasis MCX 混合型阳离子交换固相萃取小柱进行样品提取净化。

图3 20 种植物源性毒性成分过不同萃取小柱的回收率Fig.3 Recovery rate of 20 plant-derived toxic ingredients through different extraction column
2.4 净化方法的优化
2.4.1 淋洗溶剂的优化 蜂蜜属于高复杂度过饱和混合物,而糖类作为其主要化学成分,占蜂蜜干物质的95%左右,其中葡萄糖和果糖含量最高,蔗糖其次[36]。在淋洗溶剂的选择上,对甲醇和水进行考察,结果发现使用甲醇进行淋洗会导致马桑亭、闹羊花毒素V、闹羊花毒素II、闹羊花毒素III 等成分跟随淋洗液共流出,使得回收率降低。而水对糖类物质溶解性较好,且用水淋洗时目标成分随淋洗液共流出较少,故洗择水作为淋洗液。进而对淋洗溶剂的量进行了考察,分别考察了使用5、10、15 mL 水进行淋洗,结果发现15 mL 水洗脱会导致雷公藤乙素回收降低10%左右,10 mL 水能够将糖类物质洗脱完全,且目标成分获得较好的回收,回收率结果均大于70%
2.4.2 洗脱溶剂的优化 因目标成分均为生物碱类物质,在碱性条件下较易洗脱,故选择5%氨化甲醇作为洗脱溶剂。分别考察了5、9、15 mL 5%氨化甲醇3 种洗脱剂对目标分析物的影响,综合考虑目标分析物的洗脱效果和节省溶剂等因素,最后选择9 mL 5%氨化甲醇作为洗脱溶剂,能够达到最佳洗脱效果。
2.5 基质效应的影响
基质效应普遍存在于质谱检测中,表现为离子增强效应或离子抑制效应,从而导致定量结果有一定的偏差。通常基质效应消除方法有固相萃取净化、同位素内标法、稀释法等[37],而通过对比基质匹配标液与相同浓度的纯溶剂标液的仪器响应值来考察基质效应,计算公式为基质效应=基质匹配标液响应值/纯溶剂标液响应值,若两者比值在85%~115%,则基质效应可忽略[38]。本实验采用固相萃取净化的方法来消除基质效应的影响,如图4 所示,经过MCX净化之后,两者比值在85%~115%之间,其基质效应得到有效消除。因此,本文采用甲醇配制标准曲线,外标法定量。

图4 20 种植物源性毒性成分的基质效应Fig.4 The matrix effects of 20 plant-derived toxic ingredients
2.6 数据库的筛查
通过TraceFinder 软件建立的数据库与样品的保留时间、主要二级碎片、同位素分布和二级质谱图相识度比对等多种方法,综合判断,以得到准确定性结果,避免假阳性检测结果的出现。实现多组分无对照同时筛查的定性分析。具体分析物质的准确质量数及碎片离子、保留时间、电离模式如表1 所示;20 种植物源性毒性成分的提取离子流色谱图见图5。
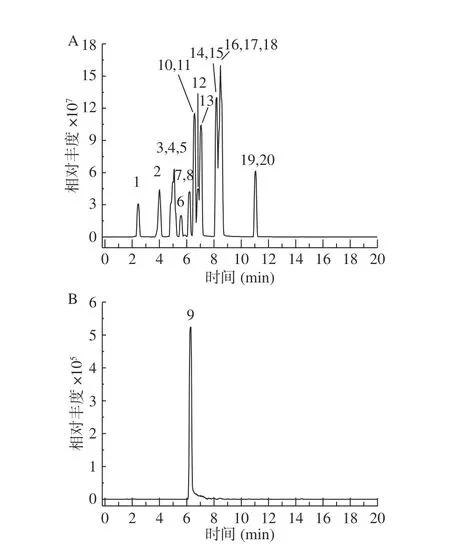
图5 20 种植物源性毒性成分的提取离子流色谱图Fig.5 Extracted ion flow chromatograms of 20 plant-derived toxic ingredients
2.7 线性关系考察
分别精密吸取“1.2.1”项下溶液0.01、0.02、0.05、0.10、0.20 mL 至10 mL 容量瓶中,使用甲醇溶液稀释成浓度约为10.0、20.0、50. 0、100. 0、200.0 μg /L的标准工作溶液。按1.2.3 方法测定。用外标法定量,以峰面积为纵坐标(y)、质量浓度为横坐标(x,μg/L)进行线性回归,求得回归方程。再分别吸取“1.2.1”溶液,逐步稀释,按照“1.2.3”色谱条件测定;取信噪比为3:1 的质量浓度为检出限,详见表2,结果表明20 种待测化合物在各自范围内呈良好的线性关系,r均大于0.995。
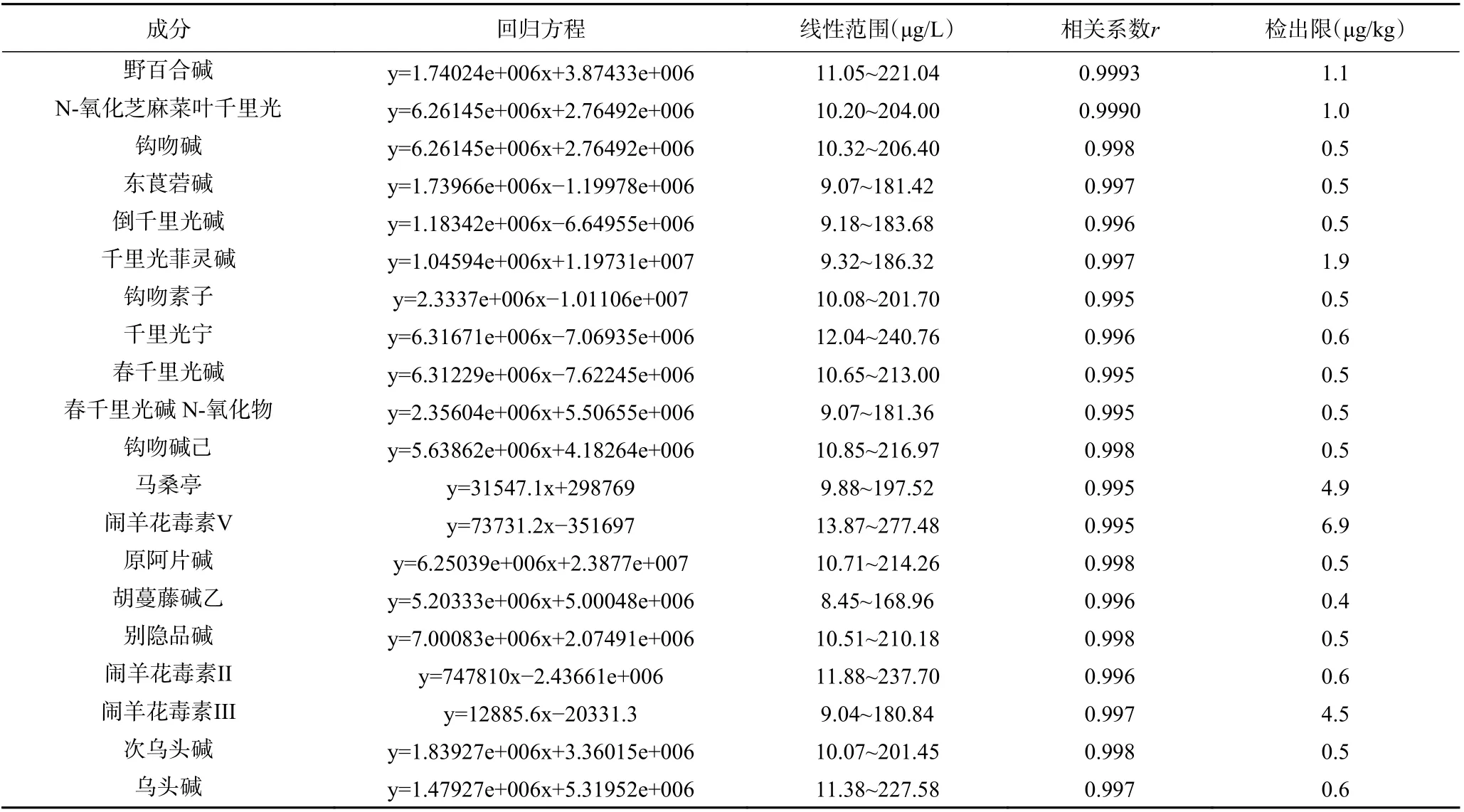
表2 20 种植物源性毒性成分的线性及相关系数和检出限Table 2 Linearities and correlation coefficients and detection limits of 20 plant-derived toxic ingredients
2.8 精密度试验
精密吸取“1.2.1”项下对照品溶液,连续进样6 次,记录峰面积,结果测得20 种化合物峰面积的RSD 范围在0.5%~2.3%之间,表明仪器精密度良好。
2.9 重复性试验
分别称取1.00 g 样品六份,置于15 mL 聚丙烯具塞离心管中,分别添加0.02 mL 的混合标准中间溶液(加入量约200 ng),按“1.2.2”项下制备并依法测定。结果测得野百合碱、钩吻碱、东莨菪碱氢溴酸盐、倒千里光碱、钩吻素子、千里光宁、钩吻碱己、马桑亭、闹羊花毒素II、闹羊花毒素III、闹羊花毒素V、原阿片碱、A-别隐品碱、次乌头碱、乌头碱、春千里光碱、春千里光碱N 氧化物、N-氧化芝麻菜叶千里光、胡蔓藤碱乙、千里光菲灵碱的RSD 分别为0.8%、 1.3%、 1.6%、 1.1%、 0.8%、 1.9%、 2.1%、1.4%、 0.8%、 0.6%、 1.3%、 0.7%、 0.5%、 2.4%、1.8%、1.6%、1.1%、0.5%、1.8%、2.0%,表明方法重复性良好。
2.10 稳定性试验
取“1.2.2”项下同一样品溶液,分别在制备后0、2、4、8、12 和24 h 按“1.2.3”色谱条件进样,记录相应的色谱峰面积,结果样品中20 种峰面积的RSD 范围在0.8%~2.1%之间,表明供试品溶液在24 h内稳定性良好。
2.11 加标回收率试验
分别称取1.00 g 样品18 份,置于15 mL 聚丙烯具塞离心管中,分别添加0.01、0.02、0.1 mL 的混合标准中间溶液各6 份,按“1.2.2”操作,进行低、中、高3 个浓度水平的加标回收试验,按1.2.3操作进样分析,计算加样回收率(结果见表3);结果如表所示各植物源性毒性成分回收率介于74.1%~114.6%之间,表明本检测方法的准确度高,可满足实验室的日常分析需求。
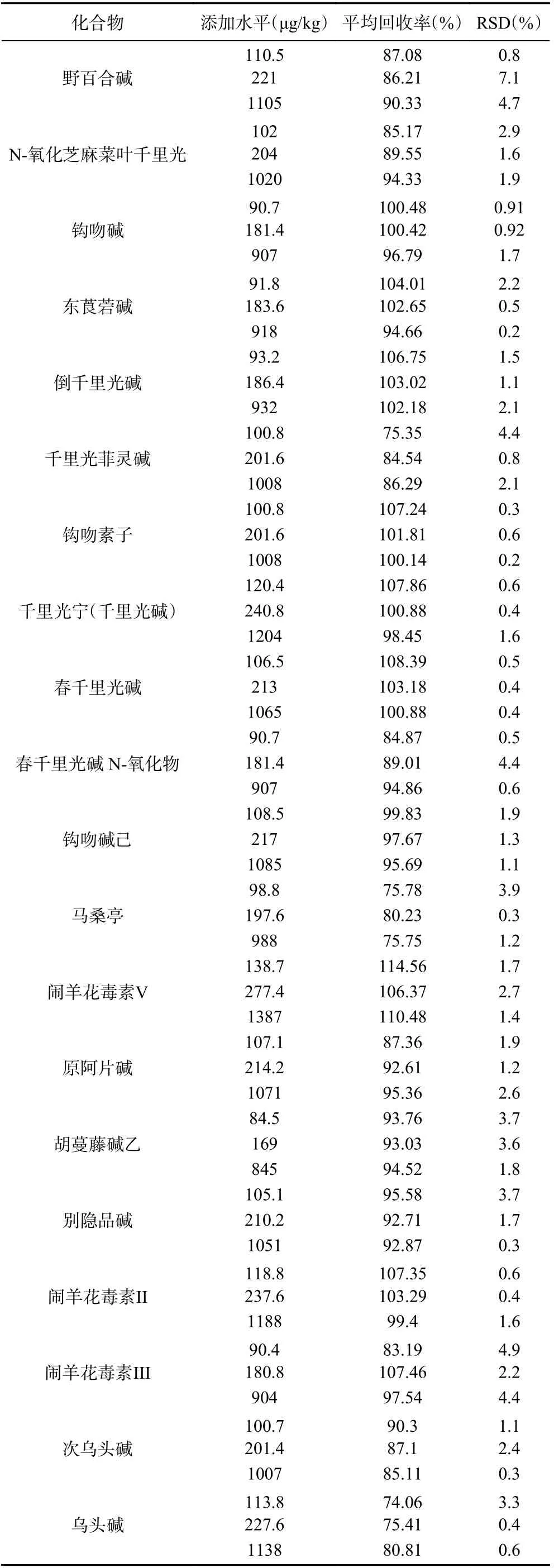
表3 20 种植物源性毒性成分的平均回收率和相对标准偏差(n=6)Table 3 Aaverage recoveries and relative standard deviations of 20 kinds of plant-derived toxic ingredients (n = 6)
2.12 实际样品分析
按“1.2”方法将66 批蜂蜜样品进行测定,结果发现在8 批蜂蜜样品中检出东莨菪碱,含量在20~1400 μg/kg 之间;其中2 批蜂蜜中另检出倒千里光碱,含量在48~150 μg/kg 之间;2 批蜂蜜中检出N-氧化千里光菲灵碱含量在49~80 μg/kg 之间,详情见表4,结果表明,在蜂蜜中检出有毒化合物占12%,在蜂蜜中存在一定的食品安全风险。
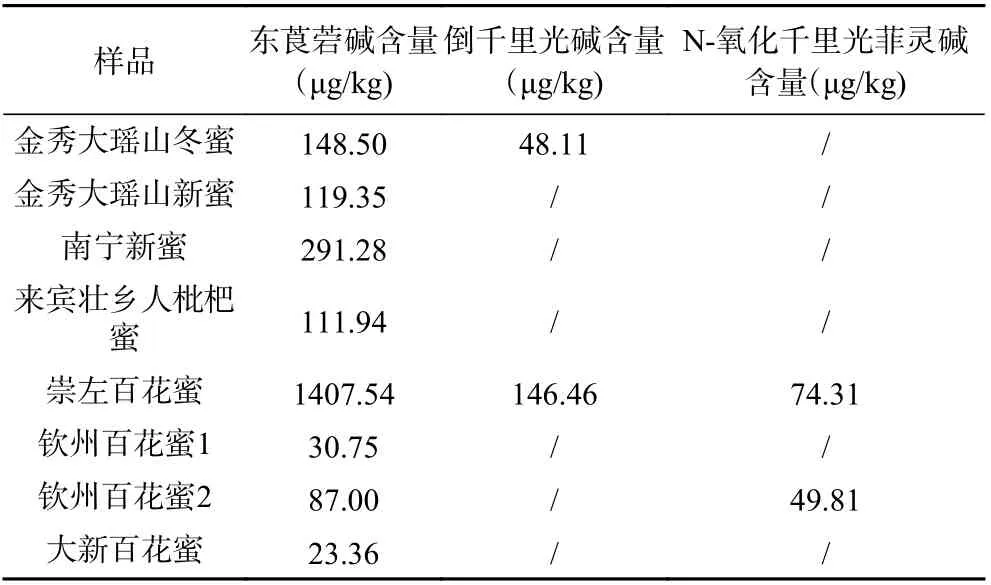
表4 样品测定结果(μg/kg)Table 4 Results of random test (μg/kg)
3 结论
本研究建立了以纯水提取,混合型阳离子交换固相萃取小柱(MCX)净化,结合超高效液相色谱-四极杆/静电场轨道阱高分辨质谱法对蜂蜜中的20 种植物源性毒性成分进行测定,其中检出了东莨菪碱、倒千里光碱和N-氧化千里光菲灵碱,提示食用蜂蜜还是存在一定的食品安全风险。方法的准确度和精密度结果都符合要求,证明该方法在对蜂蜜进行20 种植物源性毒性成分的定性定量分析时提供可靠、重复、并且准确的结果;满足日常检测和风险筛查要求。
猜你喜欢
粉末冶金技术(2021年3期)2021-07-28
人参研究(2019年3期)2019-06-26
中国中医急症(2019年10期)2019-05-21
妇女生活(2019年4期)2019-04-19
安全与环境工程(2018年2期)2018-04-13
现代养生·上半月(2016年10期)2016-10-12
中学生数理化·高二版(2016年6期)2016-05-14
中国医疗美容(2015年1期)2015-07-12
同位素(2014年2期)2014-04-16
火炸药学报(2014年3期)2014-03-20
