
Differential expression of exosomal circRNAs and regulatory framework genes in myocardial infarction patients with cardiac remodeling in response to Tongguan Capsules
2021-11-04 02:53ShuaiMaoPeipeiChenLingYuLihengGuoMinzhouZhang
TMR Modern Herbal Medicine 2021年4期
Shuai Mao, Peipei Chen, Ling Yu, Liheng Guo, Minzhou Zhang*
1 Second Clinical College, Guangzhou University of Chinese Medicine, Guangzhou 510405, China
2 Department of Critical Care Medicine, Guangdong Provincial Hospital of Chinese Medicine, Guangzhou 510120, China
Abstract Objective: Cardiac remodeling when myocardial infarction (MI) is achieved is an established prognostic factor for function-related damage and failure of hear that happen progressively.Tongguan Capsules (TGC), i.e., a Chinese herbal treatment with patent has been previously demonstrated its potential benefits in cardiac function, but little is known about related mechanisms.This study sought to isolate and characterize exosomal circRNA profiles from post-MI cardiac remodeling patients in response to TGC, and further explore the possible molecular mechanisms.Methods: Exosomes were isolated from the plasma and analyzed by the detection of protein marker expression and transmission electron microscopy.This study employed DESeq2 package within Bioconductor for exploring circRNAs and determining the circRNA with differential expressions.Co-expression investigation was performed with the use of a weighted correlation framework.An investigation was conducted on the molecular framework and channels of different circRNA based on the investigation system of the and Kyoto Encyclopedia of Genes and Genomes (KEGG) Gene Ontology (GO) channels.The prediction was conducted for circRNA-miRNA interactions on the basis of frequently employed target prediction software, and the framework was built with the use of Cytoscape software.Results: In total, 33084 circRNAs were detected in all chromosomes and 10065 circRNAs were identified with the use of circBase.Of them, 40, 207 and 258 differentially expressed circRNAs were detected between the MI group and control, MI and TGC groups, and the control and TGC groups.The differentially expressed circRNAs between the MI group and control, MI group and TGC group, and control and TGC group were significantly enriched in microtubule nucleation (BP, GO:0007020), protein binding (MF, GO:0005515), regulation of natural killer cell mediated cytotoxicity (BP, GO:0042269) corresponding to the cell cycle (hsa04110), lysine degradation (hsa00310) and lysine degradation (hsa00310) in KEGG channel investigation.Module_darkorange2 indicated the most differentiation with other modules by WGCNA investigation.This study employed a total of 14 circRNAs and 8 miRNAs which have significant interactions with each other for building the circRNA-miRNA frameworks, which indicated that has-miR-619-5p, has-miR-1268a and hasmiR-1285‐3p were under the regulation of a higher amount of circRNAs as compared with other miRNAs.Conclusion:In conclusion, different mechanisms and channels participated in the pathological processes associated with cardiac remodeling following MI.The altered circRNAs are likely to be critical to the cardioprotection of TGC through the circRNA-miRNA frameworks.
Keywords:Myocardial infarction, Exosomes, circRNAs regulatory framework, Tongguan Capsules
Background
Myocardial infarction (MI) is characterized by the irreversible myocardial necrosis caused by a prolonged interruption of oxygen supply secondary to coronary occlusion [1].MI is one of the leading causes of mortality and hospital admissions worldwide, accentuating the demand for potent approaches for the prevention and treatment of this condition [2].Practiced for thousands of years, traditional Chinese medicine (TCM) has been demonstrated to bring multiple benefits to patients suffering from MI, based upon its unique theories of etiology, diagnosis, and subsequent treatment.Sometimes TCM has been demonstrated to be a classification method in patient stratification when integrated with biomedical diagnostic methods [3].The TCM treatment for MI has been guided by diagnostics and syndrome differentiation, which are the comprehensive responses of a certain stage in the disease process [3].
From the perspectives of TCM, the key pathogenesis of MI is blood stasis due to Qi-deficiency based on physical signs and subjective symptoms of the patients [4].The validation of these observations has been supported by several epidemiological studies, including a retrospective study of 5284 patients [5].Moreover, the promotion of blood circulation by combining the supplementing Qi has become a pivotal point in TCM treatment methods for MI, established by several clinical trials with the resulting significant reductions in adverse severe vascular events [6, 7].Importantly, it has been recently established that preparation of Tongguan capsules (TGC) containing the active components of Danshen(Salvia miltiorrhiza Bge.)andHuangqi(Radix Astragalus Mongolici),induced a clinically-observed cardioprotecting effects.Recently, our published data indicated that administration of TGC could significantly mitigate the adverse post-MI remodeling [8].However, the detail physiological and molecular responses to TGC is still unclear.
Over the past few years, increasing evidence has demonstrated that the nano-sized vesicles termed exosomes may comprise fundamental properties in ischemic cardiovascular disease [9, 10].By merging their membrane contents into the recipient cell membrane and delivering effectors, including transcription factors, small and large non-coding regulatory RNAs (such as circular RNAs) and mRNAs into recipient cells, exosomes participate in pathophysiologic mechanisms [11, 12].However, exosome contents vary based upon different pathological conditions, and their differences might result in different fates of target cells.Hence, it is essential to investigate exosomes for their potential use as biomarkers for specific pathological conditions [13].Among the contents of exosomes, circular RNA (circRNA), a type of non-coding RNA that may regulate other RNA transcripts, has been shown to govern essential processes that contribute to the pathophysiological consequences of acute MI [14].Several lines of experimental evidence have demonstrated that circRNAs can block mitochondrial fission and cardiomyocyte death, and also regulate apoptosis [15, 16].Whether circRNAs derived from plasma exosomes could be used as novel biomarker, or have an essential role in the classification of MI remains undetermined.Herein, we speculated that exosomal circRNA expression profiles could serve a major parameter in post-MI remodeling patients treated with TGC.Therefore, in this study, wholetranscriptome RNA sequencing (RNAseq) was performed so as to uncover the global molecular responses to TGC at circRNA level, to furtherly revealing the underlying regulatory mechanisms.
Materials and methods
Patients and serum samples
On the whole, patients with acute MI receiving percutaneous coronary intervention identified in the Guangdong Provincial Hospital of Chinese Medicine were used in this study, including ten cases treated with just standard pharmacological therapy and the other ten cases with TGC and standard pharmacological therapy (TGC group).After a 6-month follow-up, some MI patients developed cardiac remodeling (defined as > 20% change in left ventricular end-diastolic volume index [LVEDVi] assessed by echocardiography), and were assigned into cardiac remodeling after myocardial infarction (MI) group.In addition, ten healthy volunteers with no MI and exhibiting no pathological symptoms served as controls (control group).Serum samples were obtained when 6-month follow-up after acute MI diagnosis.The informed consent was gained from all subjects in a written form.The present work received the approval from the Ethical Committee of the Guangdong Provincial Hospital of Chinese medicine (B2015-129-01).This clinical trial was registered in ChiCTR-IPR-17011618.
Isolation of exosomes
The extraction was achieved for serum from patients with MI and healthy donors on the basis of centrifugation (1500for 10 min under the ambient temperatures) soon when serum was collected and then kept under the temperature of -80°C in RNAse-free Eppendorf ™ tubes till further application.Exosomes were isolated from serum sample with the use of ExoQuick Exosome Precipitation Solution (System Biosciences, Palo Alto, CA) by complying with the guideline of the producer.In brief, thawing and centrifugation were achieved for serum sample at 3000for 15 min for discarding cell debris, following which, the introduction of 63 μL of the ExoQuick reagent and 250 μL of the serum was achieved within RNAse-free Eppendorf tube.After 12 h of incubation at 4°C, the collection was achieved for precipitated exosomes based on the centrifugating process at 1500in a period of 30 min.
Detection of exosomes
In terms of the transmission electron microscopy (TEM) morphology study, 3 μL of the exosome pellet was loaded onto formvar grids with the coating of carbon and received the incubation in a period of 5 min under ambient temperatures.Next, standard uranyl acetate staining was performed.The cleaning of grid was achieved under three variations of PBS, and the partial drying was achieved under ambient temperatures prior to the observation with the use of TEM (Hitachi H7500 TEM, Tokyo, Japan).Micrographs were used to quantify the diameters of exosomes.Western blotting was used to detect the typical exosomal makers Alix, CD63 and CD81.Briefly, the serum was processed for exosome isolation, and then the proteins were extracted from the serum and used as controls.Briefly, the loading was achieved for protein lysates (30μg) on SDS-PAGE and then the transfer was achieved to PVDF membrane.The PVDF membranes were blocked with 5% BSA and then incubated overnight with monoclonal antibodies against Alix (1:500, Abcam RayBiotech Company, Norcross, GA, the United States of America) CD63 (1:400, Abcam) and CD81 (1:2000, Abcam) solute in PBS-T.The membranes were then probed with secondary antibody (1:4000; Abcam).This study calculated the immunoreactivity with the use of improved chemiluminescence (Millipore, the United States of America).
RNA preparation, library building and RNA sequencing
The extraction was achieved for overall RNA in exosomes with the use of the TRIzol reagent (Invitrogen, Shanghai, China) in accordance with to the producer’s protocol.The RNA quality was checked with the use of Bioanalyzer 2200 system (Agilent, Santa Clara, CA, the United States of America).The quantity and integrity of overall RNA were measured with the use of NanoDrop™ ND-2000 system (Thermo Fisher Scientific, Scotts Valley, CA, the United States of America) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, the United States of America), separately.Prior to the building of RNA-seq library, a circRNA enrichment tool (Cloud-seq, the United States of America) and a Ribo-Zero rRNA removal tool (Illumina, San Diego, CA, the United States of America) were employed for removing rRNA (RIN > 8.0) and enriching circRNA.The RNA-seq library was built with the use of pretreated RNAs, and a TruSeq stranded overall RNA library prep kit (Illumina, San Diego, CA, the United States of America), by complying with the producer’s guidelines.The libraries received the denaturation to be singlestranded DNA molecule, the capture on Illumina flow cell, the amplificationin situas clusters, and lastly the sequencing for 150 cycles on an Illumina HiSeq™ 4000 sequencer (Illumina, San Diego, CA, the United States of America), by complying with the producer’s guidelines.
Differentially expressed circRNA screening and clustering investigation
Raw reads with adaptors, > 5% nucleotides not known, and bases exhibiting small quality received the removal.The mapping was conducted for qualified reads against human genome reference (GRCh37/hg19) with the use of bowtie2 and BWA [17, 18].The extraction and realignment were achieved for not mapped reads toward genome references for identifying candidate back-spliced junction read.Based on the Fragments per Kilobase Million (FPKM), the expressing state of the respective circRNA was calculated.The DESeq2 in Bioconductor was used to determine differentially expressed circRNAs, by complying with the criteria of |log2 (fold change)| > 2,P< 0.05 and a false discovery rate (FDR) < 0.05.Linear transcripts were annotated by complying with the location of the chromosome where the circRNA sequence overlapped.This study employed circBase database and circ2Trait disease database for annotating the identified circRNAs.This study employed Venn investigation for indicating the amounts of circRNAs with differential expressions among 3 compared groups.The non-supervised hierarchical clustering was illustrated as a heat map to display the expression patterns of circRNA with differential expressions.The common differentially expressed circRNAs were indicated in a Volcano Plot with the use of different colors.
Co-expression investigation with the use of a weighted correlation framework (WGCNA)
This study employed WGCNA package in R was used to construct the weighted gene co-expression framework.The gene expression matrix was imported into R version 3.5.1 (http://mirror.lzu.edu.cn/CRAN/).This study determined the strength of the Pearson link of the respective gene pair and, built an adjacency matrix through the elevation of the matrix toward a soft-threshold power with the use of the pickSoft Threshold function of WGCNA.This function provides a scale-free topology fit index, which reaches values above 0.9 for low powers ( < 30) [19].On the basis of the taken power value, this study found framework modules with mergeCutHeight of 0.25 as well as minModuleSize of 30.The respective module was assigned a specific color, and a grey color indicated genes that are outside of any module.
The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) channel investigation
GO (http://www.geneontology.org) and KEGG (http://www.genome.jp/kegg) analyses were performed for the differentially expressed circRNA-associated genes.P-value < 0.05 was considered to be significant.The top ten enriched GO terms among three comparison groups are presented.The KEGG channel investigation was adopted to determine the involvement of linear transcripts in different biological channels.The top enriched channels are also described.Based on the results of the enrichment investigation, the interaction between KEGG channels was analyzed.KEGG channel framework investigation was also conducted to explore the upstream and downstream signal channels.
Prediction of circRNA-miRNA interactions
CircRNA-miRNA interactions were predicted by popular target prediction software and the framework was built with the use of the Cytoscape software.Specific predictions were based on the miRanda, RNA22, miRWalk, Targetscan and miRDB databases.And for each circRNA, we indicated the top five miRNA that potentially bind to the circRNA, and the five most likely target genes for each miRNA.
Statistical investigation
A statistical investigation was performed with the use of Student’s t-test to compare two variables of the sequencing data.The differences with fold change (FC) ≥ 2.0 andP<0.05 were considered to be statistically significant.The false discovery rate (FDR) was calculated to assessPvalue significance.
Results
Overview of circRNA sequencing data in exosomes
TEM and Western blot investigation were employed for identifying the exosomes obtained.Under TEM, these exosomes were irregular spheres ranging of 30-100 nm in diameter, with a clearly defined and relatively intact membrane (Figure 1A).Further investigation by western blotting indicated that the expression of the typical exosomal markers Alix, CD63 and CD81 were significantly higher than that in serum (Figure 1B).These results demonstrated the exosomes isolated from serum was successful and the exosomes could be used for further study.In an initial effort to identify differentially expressed circRNAs in exosomes, we profiled the expression of circRNAs with the use of an Agilent 2100 Bioanalyzer.Clean reads ranged from 39370414 to 60548018 were obtained in exosomes for different samples, respectively (Table 1).Further investigation indicated that the mapping ratio of clean reads aligned to total reads were ranged from 51.06% to 88.44% in exosomes, respectively (Table 2).Furthermore, 33084 circRNAs with were detected in all chromosomes and 10065 circRNAs were identified in circBase (Supplemental Table 1).Of them, most of the circRNAs overlapped with known genes including 74.7% multi-exon, 16.9% exon-intron circRNA, 2.1% intronic circRNA and 4.8 single-exon circRNA, whereas smaller fractions aligned to unannotated regions (Figure 2A).And the correlation among different

Table 1.Statistical analysis for sample sequencing data evaluation

Table 2.Statistics for mapped ration between mapped reads and total reads
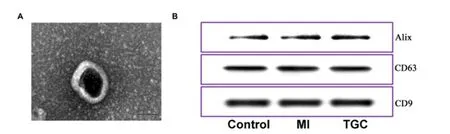
Figure 1.Detection of exosomes from serum.
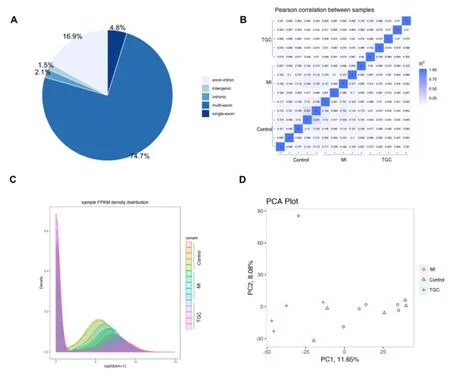
Figure 2.CircRNA identification and its expression characteristics.
samples were evaluated with the use of Pearson correlations, FPKM density distribution, and PCA investigation (Figure 2B-D).These results indicated significant differences in the correlation among different samples.
Identification of differentially expressed circRNAs
To determine the differentially expressed circRNAs in different groups, the DESeq2 package in Bioconductor was used to determine differently expressed circRNAs with a |log2 (fold change) | > 2, aP-value < 0.05 and FDR < 0.05.In total, 408 differentially expressed circRNAs were detected in the samples from different groups (Figure 3A).The expression changes of the total differentially expressed circRNAs in different groups are shown in a Venn diagram (Figure 3B).Of them, 40 differentially expressed circRNAs, 12 upregulated and 28 downregulated, were detected between MI group and control (Figure 3C and Supplemental Table 2).A total of 207 differentially expressed circRNAs, 142 upregulated and 65 downregulated, were detected between MI and TGC groups (Figure 3D and Supplemental Table 3).A total of 258 differentially expressed circRNAs, 61 upregulated and 197 downregulated, were detected between the control and TGC groups (Figure 3E and Supplemental Table 4).
The GO and KEGG investigation
The parental genes of circRNAs were obtained from the circBASE database (www.circbase.org/).To determine the function of differentially expressed circRNAs were used for GO investigation.The GO categories of molecular function (MF), cellular component (CC) and biological process (BP) for differentially expressed circRNAs were significantly enriched, respectively, in different groups and the top 10 GO terms of the differentially expressed circRNAs were listed in Table 3.Based on the GOChord plotting function, the differentially expressed circRNAs between MI group and control were significantly enriched in microtubule nucleation (BP, GO:0007020), synaptic vesicle budding from presynaptic endocytic zone membranes (BP, GO:0016185), and response to angiotensin (BP, GO:1990776) (Figure 4A).The differentially expressed circRNAs between MI and TGC groups were significantly enriched in protein binding (MF, GO:0005515), endosome to lysosome transport (BP, GO:0008333), and cytosol (CC, GO:0005829) (Figure 4B).The differentially expressed circRNAs between the control and TGC groups were significantly enriched in the regulation of natural killer cell-mediated cytotoxicity (BP, GO:0042269), the SWI/SNF complex (CC, GO:0016514), and natural killer cell activation (BP, GO:0030101) (Figure 4C).These results of GO investigation revealed the function of the differentially expressed circRNAs in MI.KEGG channel investigation was performed for further investigation of all differentially expressed circRNAs.The significantly enriched KEGG channels are listed in Table 4.The differentially expressed circRNAs between MI group and control were significantly enriched in the cell cycle (hsa04110), the reninangiotensin system (hsa04614) and glycosaminoglycan degradation (hsa00531) (Figure 5A).The differentially expressed circRNAs between MI and TGC groups were significantly enriched in lysine degradation (hsa00310), the MAPK signaling channel (hsa04010), and the VEGF signaling channel (hsa04370) (Figure 5B).The differentially expressed circRNAs between control and TGC groups were significantly enriched in lysine degradation (hsa00310), the mRNA surveillance channel (hsa03015), and the cell cycle (hsa04110) (Figure 5C).
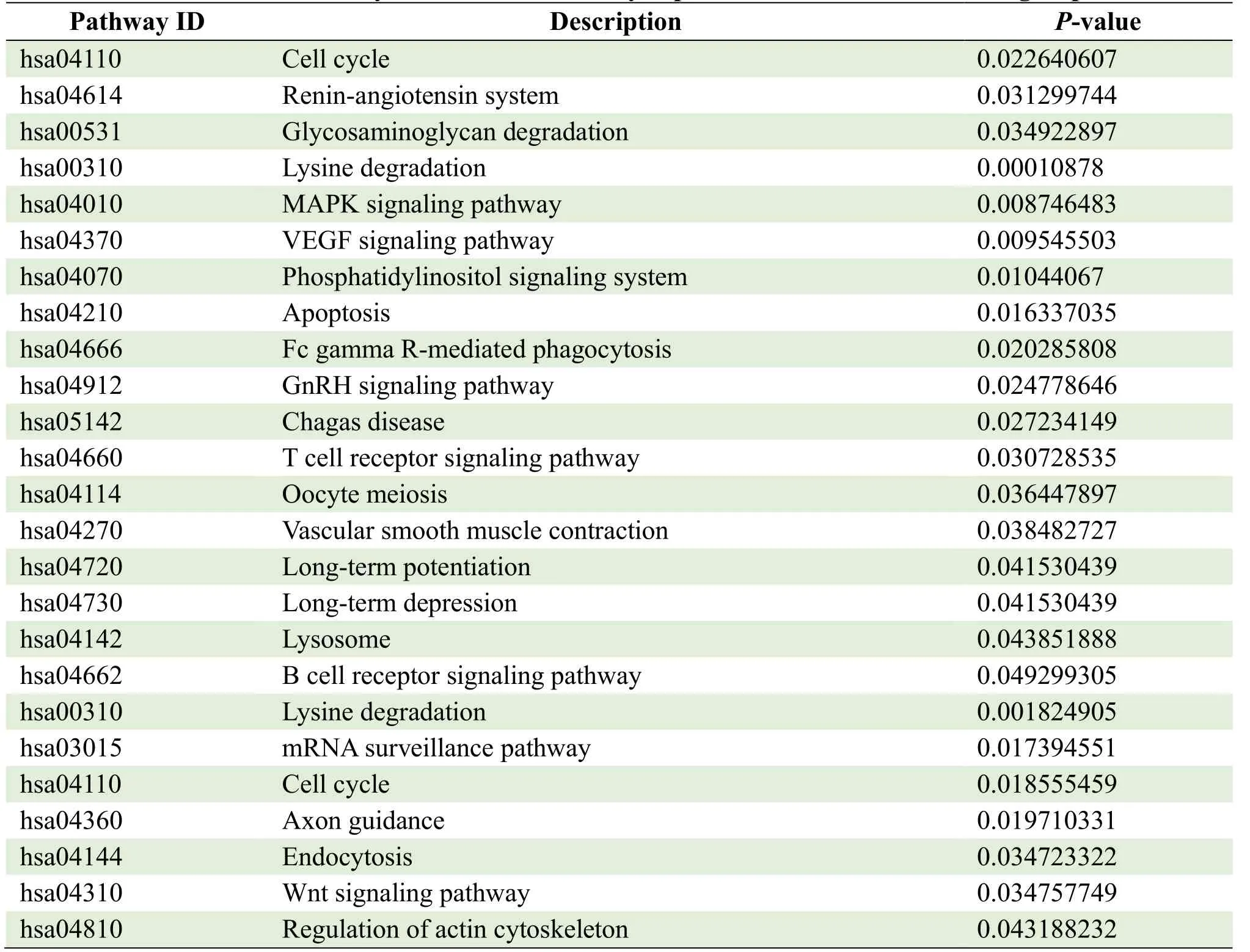
Table 4.KEGG analysis of the differentially expressed circRNAs in different groups

Figure 5.KEGG analysis of the differentially expressed circRNAs in different groups.
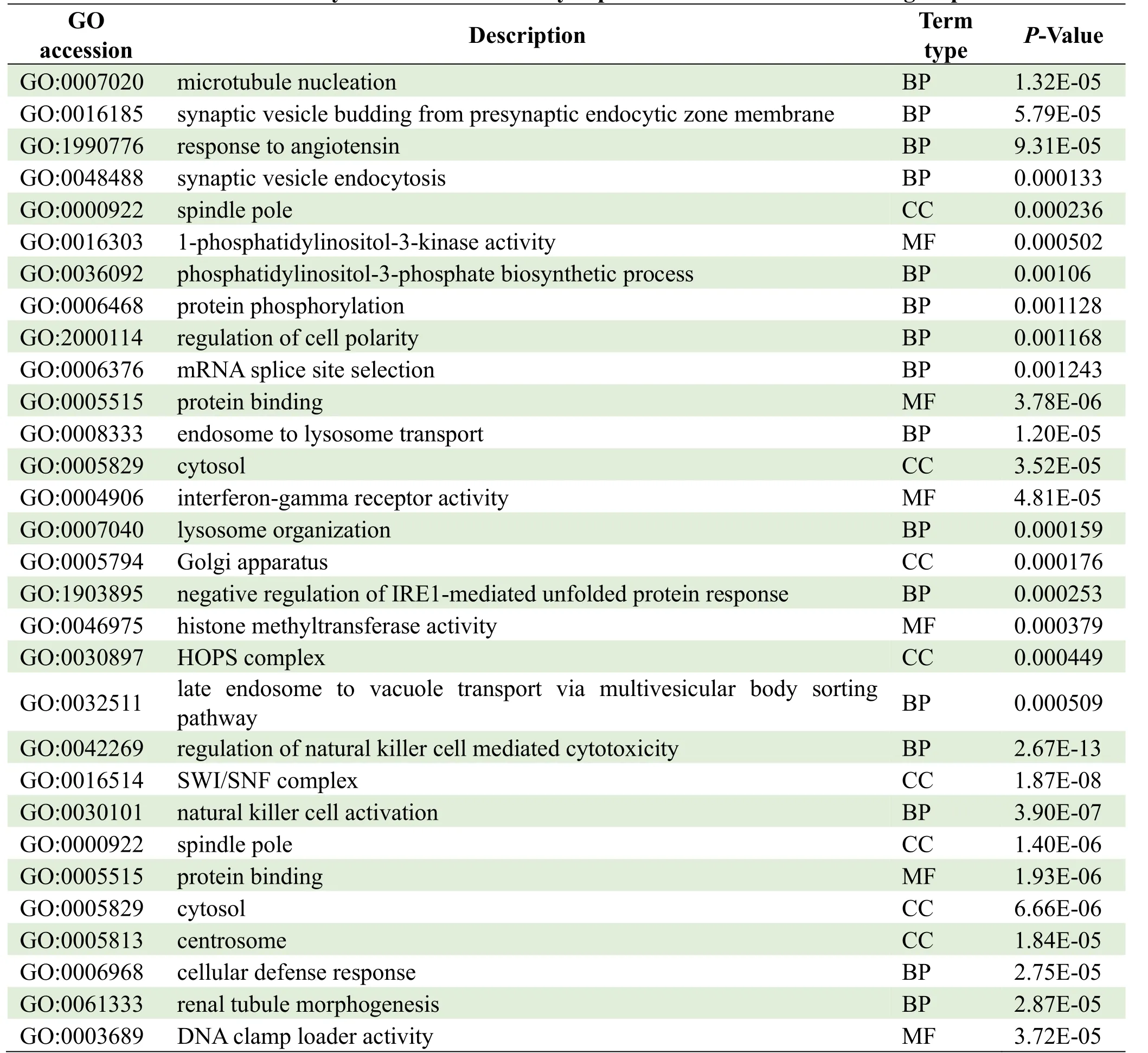
Table 3.GO analysis of the differentially expressed circRNAs in different groups
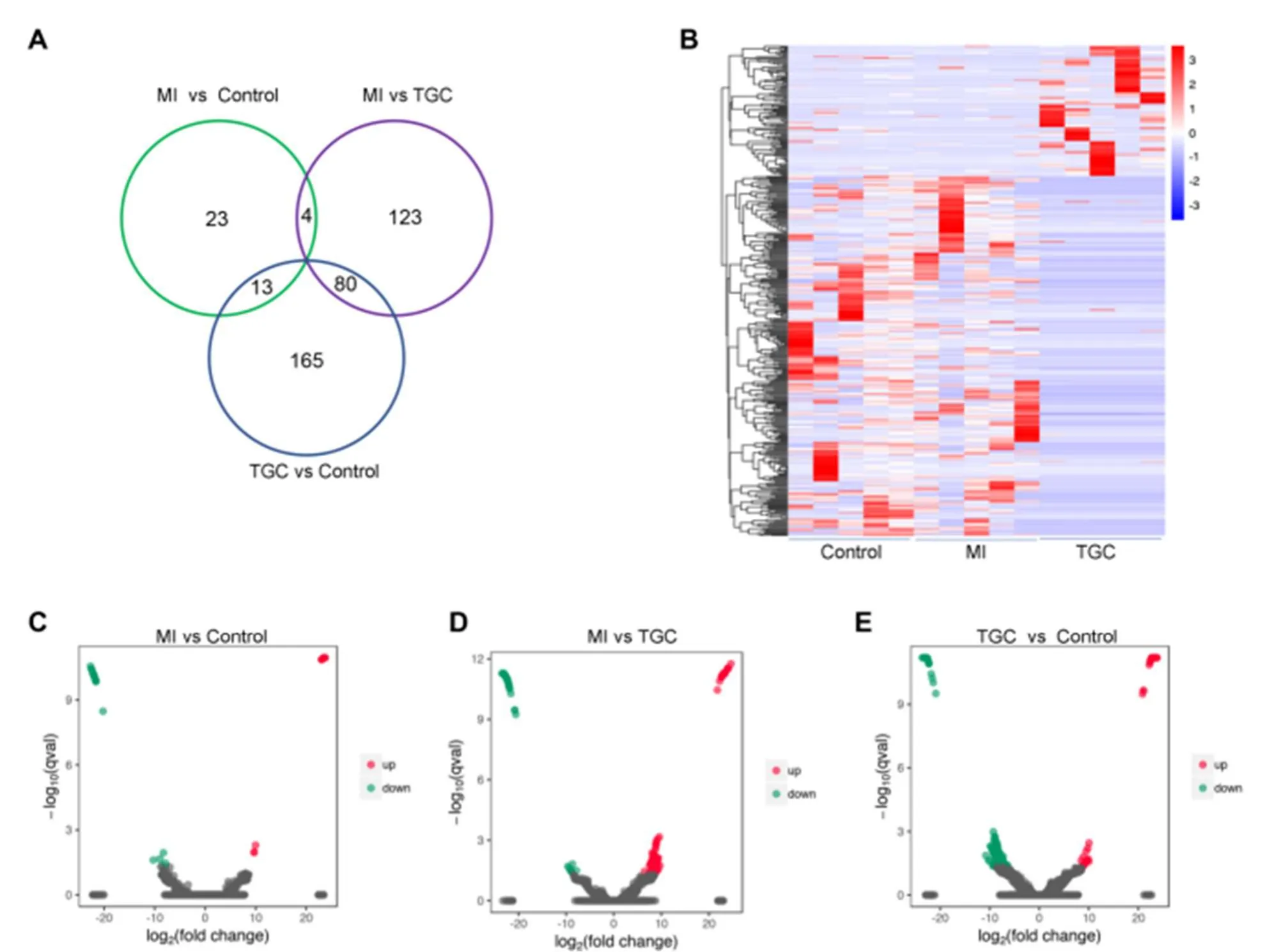
Figure 3.Overview of differentially expressed circRNAs in exosomes from different groups.

Figure 4.GO enrichment analyses of the differentially expressed circRNAs in different groups.
Co-expression investigation with the use of the weighted correlation framework (WGCNA)
A WGCNA investigation was performed based on the differentially expressed circRNAs in MI to identify gene co-expression modules that were correlated with the clinical outcomes of patients.Based on a softthreshold power of five that enabled a scale-free topology fit index above 0.73 (Figure 6A), the experimental results demonstrated that genes were clustered into 18 modules (Figure 6B and C).The correlation investigation between 18 co-expression modules is shown in Figure 6D.The results indicated that module_darkorange2 indicated the most extreme deviation from correlation with other modules, suggesting the circRNAs from module_darkorange2 may play crucial roles in MI.
CircRNA-miRNA interactions
Increasing numbers of studies have demonstrated that circRNAs could act as miRNA sponges to regulate the expressing states of other related RNAs.Therefore, it is important for us to identify the interaction of circRNAs and miRNAs.We built a circRNA-miRNA-target gene framework for differentially expressed circRNAs with the use of Cytoscape respectively.Candidate miRNAs binding of differentially expressed circRNAs are listed in Supplementary Table 5.We selected 14 circRNAs and eight miRNAs, which have significant interaction with each other, to construct the circRNA-miRNA framework (Figure 7).The framework indicated that has-miR-619-5p, has-miR-1268a, and has-miR-1285-3p were regulated by a greater number of circRNAs than other miRNAs.These results demonstrated that has-miR-619-5p, has-miR-1268a and has-miR-1285-3p may play important roles in the regulation of key circRNAs in MI.
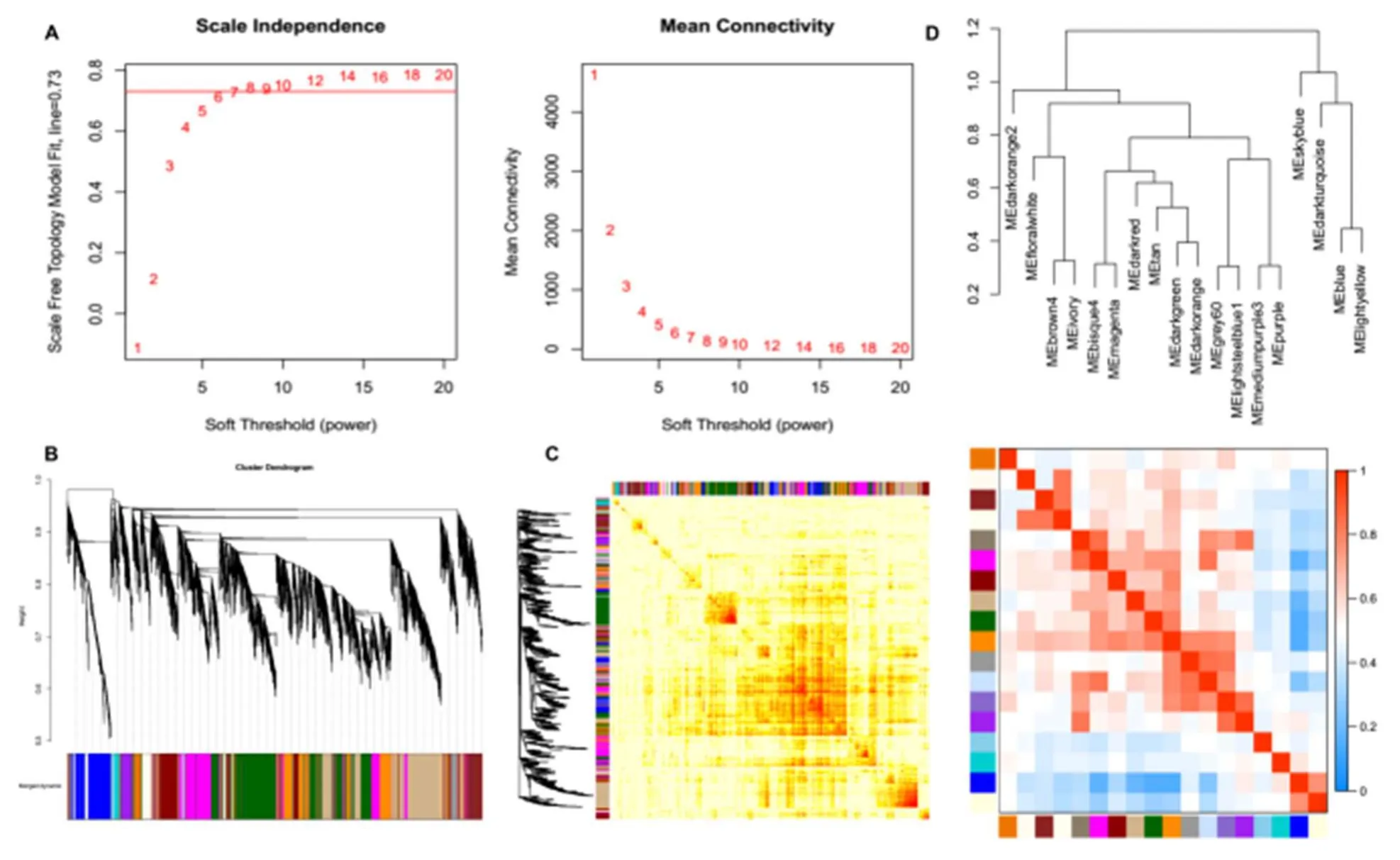
Figure 6.WGCNA analysis of the differentially expressed circRNAs in different groups.
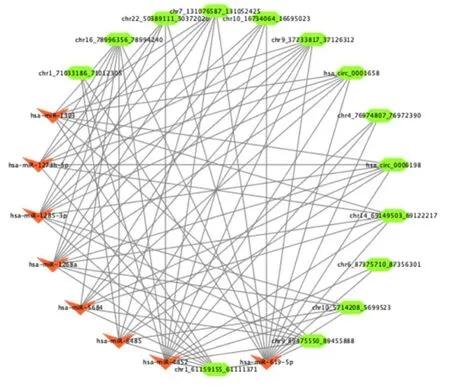
Figure 7.miRNA-circRNA regulatory interaction networks analysis.
Discussion
MI is one of the leading causes of death worldwide, as it contributes to the majority of mortality associated with coronary artery disease [20].Recent trial results show that adding daily oral TGC to the standard guidelines for basic drug therapy in patients undergoing percutaneous coronary intervention after MI significantly improved cardiac remodeling and overall clinical outcome.In the long history of clinical practice, TCM practitioners have typically classified patients of the same disease into subgroups as different syndromes guiding specific TCM treatments [21].The efficacy of the ancient “precision medicine” had been confirmed by many years of clinical use.Therefore, the mechanistic explanation of beneficial effects of TGC is pressing.Increasing evidence suggests that exosomes and circRNAs played crucial roles in cardiac remodeling after MI.However, the exosmal circRNAs in post-MI remodeling treated with Chinese medicine is less studied.Therefore, this study explored the key exosmal circRNAs in post-MI remodeling patients treated with TGC to develop a potential biological marker in the prognosis of MI patients.Exosomes are secreted by various types of cells containing with numerous paracrine factors, including proteins, lipids, and RNA.It is an important intercellular communication mediator in the heart and all major cardiac cell types can modulate recipient cellular function by releasing exosome.Exosomes have been shown to have cardioprotective effects, and participate in cardiac repair after MI, especially for those secreted by different cardiac stem cells [22, 23].For example, Bian et al., found that extracellular vesicles derived from human bone marrow mesenchymal stem cells promoted angiogenesis within a rat MI model [24]; Liu et al., indicated that exosomes derived from dendritic cells improved cardiac function via the CD4+ T lymphocytes after MI [25]; In addition, researchers have developed next-generation therapeutics with engineered exosomes for MI.Therefore, we speculated that exosomes represent a potential therapy methods for MI patients with cardiac remodeling.Thus, it is of great significance to choose exosomes from MI patients treated with TGC for study.
Increasing numbers of studies have demonstrated non-coding RNAs from exosomes played essential roles in MI with different processes.For example, Zhu et al., demonstrated that hypoxia-elicited mesenchymal stem cell-derived exosomes improved cardiac repair through miR-125b-mediated in MI [26]; Liu et al., indicated that miR-93-5p-containing exosomes treatments attenuated acute MI-triggered myocardial damage [27]; Luo et al., considered that exosomes from MiR-126-overexpressing adipose-derived stem cells were therapeutic in relieving acute MI injury [28].Ma et al., demonstrated that microRNA-132 delivered by mesenchymal stem cell-derived exosomes promoted angiogenesis in MI [29].These results provided valid evidence for the function of non-coding RNAs in MI.However, there are few miRNAs is really used for the diagnosis of MI.CircRNAs represents a novel kind of endogenous non-coding RNAs that are generated by an incorporation of the 3′ end and 5′ end non-colinear reverse splicing and can regulate gene expression at the transcriptional and post-transcriptional level [30].CircRNAs were demonstrated as ideal alternatives in terms of diagnostic biological marker and therapy target within cardiovascular diseases, as well as in MI [31, 32].Huang et al., indicated that the loss of the super-enhancer-regulated circRNA Nfix induces cardiac regeneration after MI [33].Garikipati et al., demonstrated that the circular RNA CircFndc3b modulates cardiac repair after MIvia FUS/VEGF-A axis [34].However, whether circRNAs derived from plasma exosomes serve as novel biological markers or have an essential role in the classification of MI remains undetermined.In the present study, we found that there were 23, 123, and 165 specific differentially expressed circRNAs in MI, TGC, and control group, respectively, suggesting that the expression changes of these circRNAs may represent different process in MI patients.These circRNAs may be potential biological markers for the diagnostic and prognostic of MI patients treated with or without TGC.
To further determine the regulatory role of circRNAs in MI patients treated with TGC, KEGG and GO analyses were performed for the function-related annotation of parental circRNAs.Based on the GOChord plotting function, the most significantly enriched in the microtubule nucleation (BP, GO:0007020) in MI group while significantly significant enriched in the regulation of natural killer cell-mediated cytotoxicity (BP, GO:0042269) in TGC groups when compared with control group.The microtubule nucleation function receives the mediation primarily through the capability exhibited by PCM protein for recruiting γ-tubulin ring complex (γTuRC), which in turn serve as templates for the growth of microtubules.Microtubule trafficking is a key process that plays crucial roles in the development of MI related to cardiomyocyte dedifferentiation and proliferation [35], and autophagy [36].Fang et al.found that systemic inflammatory response following acute MI is related to natural killer cell-mediated cytotoxicity [37] (Systemic inflammatory response following acute MI).Gao et al.also demonstrated that natural killer cellmediated cytotoxicity was significantly enriched in genes with differential expressions related to acute MI [38] (Bioinformatics analyses of genes with differential expressions related to acute MI).These results of the GO investigation of the differentially expressed circRNAs revealed that different regulatory mechanisms were involved in MI.KEGG channel investigation was performed to analyszs all differentially expressed circRNAs further, and the significantly enriched KEGG channel investigation also confirmed these results.KEGG channels investigation indicated that it was significantly enriched in cell cycle in MI group compared with control group.These results are consistent with previous reports that acute MI-triggered functionrelated cardiomyocytes to re-enter the cell cycle and regulation of the cell cycle could stimulate adult cardiomyocyte proliferation and cardiac regeneration [39, 40].However, the differentially expressed circRNAs in TGC were significantly enriched in lysine degradation, which contributes to natural killer cellmediated cytotoxicity [41].These results provide a solid foundation for revealing the biological processes and mechanisms involved in the development of MI treated with TGC.
The classic channel is the circRNA-miRNA-mRNA channel, which indicates that the circRNA binds and suppresses miRNA, thereby exerting an effect on the target mRNA expressing state [30].Studies also indicated this regulatory framework in MI.Recently, Geng et al.demonstrated that the circRNA cdr1as facilitates MI through the mediation of mir-7a regulation on its target gene expressing state [42].Gan et al.indicated that the circular RNA_101237 mediates anoxia/reoxygenation injury by targeting let‑7a‑5p/IGF2BP3 in cardiomyocytes.Cai et al.found that the circular RNA Ttc3 regulates cardiac function after MI by absorbing miR-15b.According to the mentioned finding, circRNA is capable of binding to miRNA and controlling gene expressing state at the posttranscriptional level, thereby causing a novel knowledge dimension about MI pathogenesis.To determine the potential regulatory framework in MI, a competing endogenous RNA (ceRNA) framework was built.We determined 14 circRNAs and 8 miRNAs which indicated more significant interactions with each other to build the circRNA-miRNA framework.The framework indicated that has-miR-619-5p, has-miR-1268a, and has-miR-1285-3p were controlled by a higher number of circRNAs compared to other miRNAs.These results demonstrated that has-miR-619-5p, has-miR-1268a, and has-miR-1285-3p might play an essential role in MI regulation by circRNAs.As many other novel efforts, our exploratory trial might also have certain limitations, due to the fact that the reported results without further validations.Therefore, a future more comprehensive work is needed to confirm our exploratory.
Conclusions
In this study, we compared the molecular frameworks of post-MI remodeling patients treated with TGC and analyzed their shared and differing circRNAs mechanisms.Our results help develop circRNA to be a novel class of exosome-related biological markers, and suggest a probably biology function in terms of exosomal circRNA.A better understanding of the essences of the biological characteristics of cardiac remodeling after MI is beneficial for exploring different therapies to enhance the effectiveness and pertinence of treatment.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and comparable or amendments ethical standards.
Authors' contributions
SM and MZZ designed the research.SM drafted this manuscript; SM, PPC, LY, and XJZ carried out the experiments, LHG made statistical analysis; MZZ revised the manuscript and contributed to the rationalization of the study.All authors read and approved the final manuscript.
Acknowledgements
Gratefully acknowledge the contributions of all staff for their participation in the study.
Supplementary materials
Supplementary data are available online atTMR Modern Herbal Medicine.
 TMR Modern Herbal Medicine2021年4期
TMR Modern Herbal Medicine2021年4期
- TMR Modern Herbal Medicine的其它文章
- Research progress on mechanism of Chinese material medica in preventing and treating insulin resistance and type 2 diabetes mellitus
- lncreased macrophage inflammation response in pancreatic cancer patients with a diagnosis of Shi-Re
- Experimental study on the effect of Huangqi and Danshen ultramicro gel on vascular healing of wound in rats based on network pharmacology
- The clinical efficacy of traditional Chinese medicine in the auxiliary treatment of grade 1 hypertension: A systematic review and metaanalysis
- Network pharmacology-based analysis on bioactive compounds and mechanisms in Yiqifumai formula in the treatment of heart failure
- Application of Chinese Herbal Medicine in the lnhibition of Salmonella and its virulence