
20号染色体的非整倍体及单亲二体的相关研究进展
2021-11-06 02:40刘勉张锐何怡周玉侠刘思平许育双王克贾蓓
中国产前诊断杂志(电子版) 2021年3期
刘勉 张锐 何怡 周玉侠 刘思平 许育双 王克 贾蓓*
(1.南方医科大学南方医院妇产科生殖医学中心,广东 广州 510515;2.深圳市宝安区妇幼保健院产前诊断科,广东 深圳 518133;3.东莞市妇幼保健院医学优生遗传科,广东 东莞 523000;4.山东省妇幼保健院产前诊断中心,山东 济南 250014;5.南方医科大学南方医院妇产科产前诊断与遗传病诊断中心,广东 广州 510515)
20号染色体属于F组的中着丝粒染色体(F组:19、20号染色体),全长共约63Mb(hg19),约含6300万个碱基对,大概占人类染色体组总DNA的2%,携带881个编码基因,其中OMIM基因435个,致病性OMIM基因114个(https:∥decipher.sanger.ac.uk/)。本综述将对20号染色体非整倍体、20号染色体单亲二体的发生机制、发生率、临床表型、治疗、预后以及再发风险进行归纳,以期对相关疾病的遗传学诊断以及遗传咨询提供参考。
1 20号染色体的非整倍体
1.1 20号染色体短臂部分单体综合征 完全型20号染色体单体,通常于孕早期流产,暂未检索到存活个体的报道。20号染色体部分单体综合征的报道主要以短臂部分单体为主,长臂部分单体报道极少。20短臂(p)部分单体综合征最早于1976年由Kalousek等[1]发现,已报道的核型包括46,XX,20p-;46,XY,del(20)(p11);46,XY,del(20)(p11p13)等。20p单体综合征主要临床表现包括低出生体重儿,精神运动发育迟缓及特殊面容等,如前额前凸、枕骨扁平、颅缝增宽、眼角错位、塌鼻梁、高腭弓、长人中、嘴角下翻、耳部外观异常、颈短而宽、脊柱侧凸、通贯掌、指(趾)畸形等。患者通常合并肌张力低、心血管系统异常等。20p单体综合征患者通常在婴幼儿期死亡[2-4]。
1.2 20-三体综合征(trisomy 20,T20)及嵌合三体
1.2.1 发生机制及发生频率 完全型T20是一种非常罕见的染色体畸变,主要由第一次减数分裂后期同源染色体不分离或者第二次减数分裂后期姐妹染色单体不分离所致。嵌合型T20在产前诊断中相对常见,通常因胚胎体细胞增殖过程中发生有丝分裂不分离导致部分细胞系形成三体,部分细胞系维持正常二倍体从而形成嵌合;也可能在配子形成过程中因减数分裂不分离形成完全型三体,但胚胎的部分三体细胞发生三体自救(trisomy rescue),丢弃一条染色体后恢复成正常的二倍体细胞,从而形成三体细胞与正常二倍体细胞的嵌合。
完全型T20是一种极为罕见的致死性染色体畸变,目前仅报道6例早期妊娠行产前诊断确诊为完全型20号染色体三体的宫内活胎病例,这些胎儿均伴有不同程度的生长发育异常[5]。完全型T20多在羊水细胞中检出,少部分在绒毛中被检出,仅有1例从产前脐血中检出完全型T20的报道[6]。嵌合型T20是产前诊断中常见的嵌合类型之一,发生率约1/7000,占所有产前诊断发现嵌合体病例的16%。约90%~93%的嵌合型T20胎儿无异常表型,约6.5%的胎儿出生后合并异常表型,因此临床工作中对于嵌合型T20的遗传咨询非常困难[5]。
1.2.2 临床特征 20号染色体三体综合征最早于1976年由Pan等[7]报道,都源自新发变异。因其不具备生存能力,完全型T20罕见,目前已报道的核型主要为47,XX(XY),+20;46,XX/47,XX,+20;46,XY/47,XY,+20[8,9]。T20主要临床表现为先天性多发畸形、患儿伴随严重消化系统异常、脊柱异常、外眦上斜、内眦赘皮等[8-10]。
20p三体综合征于1974年由Subrt等[11]首次发现,几乎都源自携带者。Subrt等报道20p三体综合征表型主要包括前额倾斜、枕骨扁平、圆脸、脸颊突出、眼眶凹陷、眼角发育不良、睑裂上斜而短、短鼻、鼻尖前倾、牙齿异常、小颏、大耳、后旋耳及低位耳,同时合并其他系统异常,如脊柱异常、先天性心脏病、肾脏异常等。患者行为粗暴、语言异常,合并中度智力障碍[12-17]。2019年Stembalska等[18]报道诊断为20号染色体部分三体(20p11.21至20q11.23)的患儿,存在一系列相似的异常表型,主要表现为发育迟缓、运动协调性受损、智力低下、言语迟缓、椎骨发育异常(椎骨融合、椎间隙缩小、脊柱裂、脊柱侧凸和/或后凸)、肾脏发育异常(肾发育不良、重复肾、肾积水、多囊肾、异位肾)以及生殖器发育不良(尿道下裂或隐睾)。其表型分析还有待更大样本量的长期随访研究。
嵌合型T20在产前诊断中较常见[5],迄今为止约有40例嵌合型T20活产胎儿的相关报道,出生后临床表型各异。Montplaisir等[5]总结所有关于嵌合型T20的临床表型如下:精神运动发育迟缓(27%)、肌张力减退(18%)、低位耳(14%)、下颌后缩(14%)、小下颌畸形(14%)、室间隔缺损(14%)、伊藤黑色素沉着症(14%)、鼻骨发育不良(10%)、扁平鼻(10%)、内眦赘皮(10%)、斜肩(10%)、椎骨发育异常(椎骨融合、椎间隙缩小、脊柱裂、脊柱侧凸和/或后凸)(10%)、屈指畸形(10%)、小头畸形(10%)、隐睾(10%)、招风耳(5%)、唇腭裂(5%)。在出生后才确诊为嵌合型T20的患儿中,部分表现为眼压降低、轻度生长发育迟缓、特殊面容及小头畸形等异常表型[19]。
1.2.3 实验室检查 产前绒毛、羊水、脐血细胞,产后胎盘、脐带以及皮肤、肾脏等组织都可行核型分析确定嵌合比例。极少通过血液细胞检出T20,目前仅报道1例在血液细胞中检出完全型T20,但仍需通过临床表型验证。由于存在限制性胎盘嵌合(confined placental mosaicism,CPM)的可能,产前绒毛活检提示嵌合型T20时应进一步行羊膜腔穿刺以排除CPM[20]。采集到羊水样本后可采取培养羊水细胞收获核型、未培养羊水细胞单核苷酸多态性微阵列(single nucleotide polymorphism array,SNP array)和羊水细胞间期荧光原位杂交(fluorescent in situ hybridization,FISH)等的联合检查。针对产前发现嵌合型20染色体三体的胎儿应进行详细的超声评估,嵌合比例较低时,产前超声可能无异常表现。在产前绒毛标本中检出为20-三体的病例中,羊水染色体检测最终确诊胎儿20-三体的比例为11.8%[21]。根据国内4家医院的统计(见表1),完全型T20及嵌合型T20在产前及流产组织标本中的总检出率约为0.05%。在所有的产前诊断标本中(包括羊水及绒毛),20嵌合三体的检出率约为0.02%(5/25 528),产前标本中尚未发现20-三体,可能由于致死性异常导致胚胎早期流产,故只能在流产组织中检出。
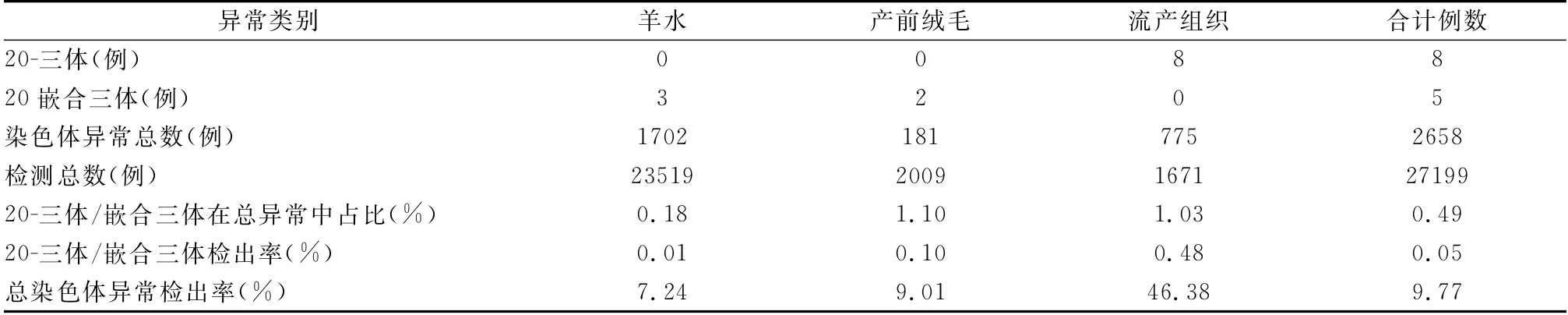
表1 27 199例产前及流产组织标本中染色体20号三体及嵌合三体的检出率*
1.2.4 治疗与预后 嵌合型T20患儿临床异质性强,治疗方案须个体化选择,若合并发育迟缓或语言障碍可行针对性康复治疗及语言训练。因90%~93%产前诊断为嵌合型20号染色体三体的患儿可无异常表型,需综合分析各种因素后,如是否为CPM、嵌合比例、胎儿超声结构筛查及是否存在单亲二体(uniparental disomy,UPD)等,进行细致评估,给予相应的咨询。
1.2.5 再发风险评估 目前产前检出的嵌合型20号染色体三体多为散发病例,再发风险极低。
2 20号染色体单亲二体
2.1 单亲二体的概述 单亲二体(uniparental disomy,UPD)指体细胞染色体核型中,同源染色体或者同源染色体上的部分片段均来自同一个亲本,而完全缺乏另一个亲本的染色体[22]。自从1988年有学者首次报道UPD导致的相关疾病以来,随着检测手段的发展,UPD所引发的疾病受到越来越多的报道与关注[23]。
UPD的发生机制包括但不限于三体自救、单体复制、配子互补及体细胞有丝分裂异常等。根据同源染色体来源,可将其分为母源性UPD(maternal UPD,mat UPD)和父源性UPD(paternal UPD,pat UPD),研究显示mat UPD的发生率是pat UPD的3倍。根据所涉及染色体的范围,又可将UPD分为涉及非整条染色体的节段性UPD、整条染色体UPD和伴有同源性UPD和异源性UPD的复杂型UPD[22]。
UPD与多种疾病相关,机制包括:①常染色体隐性基因纯合突变所导致的隐性遗传病;②基因印迹障碍;③胎盘与胎儿嵌合体导致发育障碍[23]。关于UPD的报道几乎涉及所有染色体,现我们针对20号染色体UPD进行相关综述。
2.2 父源性与母源性20号染色体单亲二体与疾病Geneimprint网站(http:∥www.geneimprint.com/site/genes-by-species)目前已收录17个位于20号染色体的印迹基因,其中12个为明确的印迹基因(imprinted),4个为很可能有印迹功能(predicted)的基因,另外1个基因HM13是否为印迹基因需进一步明确。在所收录的17个印迹基因中,12个为父源性表达,分别为ISM1、PSIMCT-1、BLCAP、NNAT、MCTS2、GDAP1L1、SGK2、L3MBTL、SANG、GNASAS、MIR298和MIR296;2个为母源性表达,分别是COL9A3和C20orf20。
2.2.1 父源性单亲二体 截至2020年2月28日,UPD网站(http:∥upd-tl.com/)共收录了7例核型正常的父源性20号染色体单亲二体[parental UPD(20),pat UPD(20)]的病例报道。父源性20号染色体单亲二体与假性甲状旁腺功能减退综合征(pseudohypoparathyroidism 1B,PHP1B)密切相关[24]。2001年Bastepe[25]等首次在1例PHP1B患者的核型中发现存在部分性pat UPD(20),随后陆续病例报道亦均证实pat UPD(20)与PHP的相关性。pat UPD20导致PHP1B的发生与印记基因GNAS(OMIM#603233)表达紊乱有关[26]。Eduardo Ferna′ndez-Rebollo等[27]描述了4例部分性pat UPD(20)的PHP1B患者,其中1例为整条长臂pat UPD(20),2例为部分性长臂pat UPD(20)(20q13.13-qter),1例为节段性异源pat UPD20,上述结果提示20号染色体有丝分裂重组也会导致UPD的发生从而引发PHP1B,进一步揭示了pat UPD(20)与PHP1B的相关性。2019年Colson等[28]利用SNP芯片比较基因组杂交技术筛查33例散发性PHP1B患者中pat UPD(20)的检出率,结果确认了6例pat UPD(20),并且这些患者的20号染色体存在典型的父源性甲基化表征,包含GNAS A/B:TSS-DMR位点甲基化的丢失,GNAS-AS1:TSS-DMR、GNAS-XL:Ex1-DMR位点甲基化的显著降低以及GNAS-NESP:TSSDMR位点高度甲基化等,提示pat UPD(20)是导致散发PHP1B最可能的原因之一,因此Colson认为若在遗传咨询中接诊偶发性PHP1B患者,pat UPD(20)应作为一项重要筛查指标。最近Gaudio等[29]进一步提示,建议对临床上出现GNAS位点DMRs甲基化异常,但染色体核型、染色体微阵列分析(chromosomal microarray analysis,CMA)结果正常的PHP1B患者行pat UPD(20)的确诊检查,确认是否存在pat UPD(20)存在。此外,Hye-Sun Park等还报道了一例合并骨肉瘤的PHP1B患者,认为骨肉瘤也与其pat UPD(20)相关[30],但仅此一例报道,需针对更多合并PHP1B的骨肉瘤患者进行pat UPD(20)检测,来明确pat UPD(20)与骨肉瘤发生之间的关联性。
2.2.2 母源性单亲二体 截至2020年2月28日,UPD网站(http:∥upd-tl.com/)共收录了13例核型正常的母源性20号染色体单亲二体[maternal UPD(20),mat UPD(20)]的病例报道。母源性20号染色体单亲二体(Mulchandani-Bhoj-Conlin综合征)是一种十分罕见的疾病,文献报道的患者不足20人,主要表现为胎儿宫内生长受限以及出生后生长发育异常。CMA检测发现mat UPD(20)发生主要是由于第二次减数分裂错误或者合子有丝分裂异常导致[29]。1999年Chudoba等[31]首次在1例重度发育迟缓的4岁男孩中检测到母源性20号染色体单亲二体。2001年Eggermann等[32]再次在1例重度发育迟缓的男婴中检测出mat UPD(20),提示mat UPD(20)可能是导致子代生长发育迟缓的原因之一。2016年Mulchandani等[33]对8例mat UPD(20)患儿的临床表型进行分析,发现所有患儿产前均合并重度宫内胎儿生长受限,出生后均出现喂养困难、发育迟缓、身材矮小等,提示早发性孤立性生长发育迟缓的患者中,mat UPD(20)应作为一项重要检测指标。因此,当临床出现生长迟缓、吞咽困难并且伴随SNP检查20号染色体上出现纯合片段(runs of homozygosity,ROH)的患者,建议对其进行mat UPD(20)的确诊检查[29]。进一步研究发现超过5%的Silver-Russell综合征及不明原因的小于胎龄儿均可以用mat UPD(20)解释,另外由于mat UPD(20)与激素受体超敏反应密切相关,可导致高钙血症与甲状旁腺激素水平降低,上述机制与mat UPD(20)致病性可能存在密切关联[34]。
2.3 UPD(20)导致疾病的特征 父源性20号染色体单亲二体与1B型假性甲状旁腺功能减退症密切相关,约18%的PHP1B患者由patUPD(20)引起[28]。假性甲状旁腺功能减退症(pseudohypoparathyroidism,PHP)是一种罕见的遗传性内分泌疾病,以低钙血症、高磷酸盐血症和终末器官对甲状旁腺激素(parathyroid hormone,PTH)的抗性为临床表现。PHP包含多种亚型,发病机制各不相同,临床表现多样,但大多具有类似的典型表型:肥胖、圆脸、身材矮小、短指、皮下骨化和智力低下等[35]。此外,Charles P等报道pat UPD(20)患儿存在明显的异常表型:小儿畸形/无耳畸形、小脑畸形、先天性心脏病、神经元室管膜下灰质异位以及结肠无神经节细胞增多症等[36]。母源性20号染色体单亲二体(Mulchandani-Bhoj-Conlin综合征)与胎儿宫内生长受限、出生后身材矮小、喂养困难以及发育缓慢密切相关[33]。
2.4 实验室检查 对UPD(20)的实验室检测,可使用甲基化多重连接探针扩增技术(methylationspecific multiplex ligation-dependent probe amplication,MS-MLPA),也可使用含SNP探针的微阵列芯片技术或高通量测序技术。
2.5 治疗与预后 携带UPD(20)的患者临床表型各异,预后也不相同,应根据患者临床表型制定个体化治疗方案,对症治疗。
2.6 再发风险评估 UPD(20)属于新发染色体变异,再发风险极低。评估UPD(20)胎儿的预后复杂,需综合胎儿超声结构筛查、UPD种类、是否存在嵌合型20号染色体三体细胞等因素细致评估。如果产后胎儿伴随PHP,需考虑pat UPD(20)的可能。
猜你喜欢
中华医学图书情报杂志(2022年1期)2022-11-18
中国现代医生(2022年21期)2022-08-22
理财周刊(2022年4期)2022-04-30
农村科学实验(2022年2期)2022-03-12
云南医药(2021年6期)2022-01-08
三农资讯半月报(2020年2期)2020-03-09
好日子(2019年4期)2019-05-11
中国实用医药(2016年36期)2017-06-20
课程教育研究·下(2016年7期)2016-08-23
海峡姐妹(2016年5期)2016-02-27
