
HPLC-MS/MS测定动物源性食品中利巴韦林和金刚烷胺药物残留
2021-11-06 13:12虞长贵
现代食品 2021年17期
◎ 虞长贵
(郑州中检科测试技术有限公司,河南 郑州 450000)
目前,利巴韦林和金刚烷胺均为动物源性食品药物残留检测项目要求,同一种样品中都要进行检测,现有检测标准是独立进行检测[1-5]。本研究使用高效液相色谱质谱联用仪同时检测动物源性食品中利巴韦林和金刚烷胺药物残留,操作简单,结果准确可靠,节约资源,为动物源性食品中利巴韦林和金刚烷胺药物残留检测提供一种检测技术手段,在实验室推广和应用方面具有重要意义[1-5]。
1 材料与方法
1.1 仪器与设备
超高效液相色谱-串联三重四级杆质谱仪(岛津公司LCMS 8050);氮吹浓缩仪(EVA32);高速离心机(日立CR21N);Milli-Q超纯水装置(Milli-Q Integral 5);PH计;分析天平(0.000 1 g);电子天平(0.001 g);振荡器(TAITECA);固相萃取装置(色谱科);超声仪;干燥箱。
1.2 材料与试剂
从市场随机采购动物源性样品。
甲醇(色谱级)、乙腈(色谱级)、甲酸(色谱级)、乙酸铵(色谱级)、三氯乙酸、氨水、酸性磷酸酯酶、混合型离子交换固相萃取小柱(CNWBOND PBA)、超纯水、利巴韦林、利巴韦林内标C5、金刚烷胺、金刚烷胺内标D6及离子型亲水性T3色谱柱(ACQUITY UPLC HSS T3 100×2.1 mm,1.8 μm)。
1.3 样品前处理
1.3.1 提取
称取5 g(精确到0.01 g)样品置于50 mL高速离心管,加入15 mL三氯乙酸溶液和2.5 mL乙腈溶液,涡旋混匀,振荡10 min,超声5 min,以20 000 r·min-1高速离心5 min,取上清液至25 mL容量瓶,样品再加10 mL三氯乙酸溶液,重复提取,合并提取液至容量瓶,定容至25 mL。
1.3.2 酶解
移取5 mL提取液至50 mL高速离心管,加入1.0 mL乙酸铵溶液,加入100 μL酸性磷酸酯酶,涡旋混匀,37 ℃干燥箱中酶解2 h。取出冷却至室温,用氨水调节pH至8.5±0.1,涡旋混匀,20 000 r·min-1离心5 min,取上清液备用。
1.3.3 净化
上清溶液转入经活化的PBA固相萃取小柱,依次用5 mL 10%乙腈-乙酸铵缓冲液、2 mL 5%氨水甲醇淋洗,弃去流出液,用真空泵抽干固相萃取柱5 min,再用4 mL甲酸-水-甲醇溶液(2∶8∶90)洗脱至氮吹管,45 ℃水浴下氮吹至近干,加入1.0 mL乙腈-水(9∶1)溶解氮吹管中残渣。经0.22 μm有机滤膜过滤,供液相色谱-质谱联用仪测定。
1.4 仪器条件
1.4.1 液相色谱条件
色谱柱:离子型亲水性色谱柱ACQUITY UPLC HSS T3 100×2.1 mm,1.8 μm;流速:0.4 mL·min-1;柱温:30 ℃;进样体积:2 µL;流动相A:乙腈;流动相B:5 mmol·L-1乙酸铵水+0.1%甲酸。
1.4.2 质谱条件
离子化模式:电喷雾电离正离子模式;检测模式;多反应监测(MRM)
2 结果与分析
2.1 提取条件选择
常用提取溶剂有甲醇、乙腈、丙酮、乙酸乙酯等试剂,拟选择甲醇、乙腈、丙酮、乙酸乙酯作为提取液。甲醇、丙酮为亲水性溶剂,不利于消除基质中一些水溶性成分。本方法考察乙腈和乙酸乙酯两种溶剂对基质进行提取,提取效果见表1。结果表明,乙腈对本方法中检测两种物质有较高的提取效果。考虑到基质中含有蛋白质等干扰物质,因此本方法采用三氯乙酸-乙腈对基质进行提取。
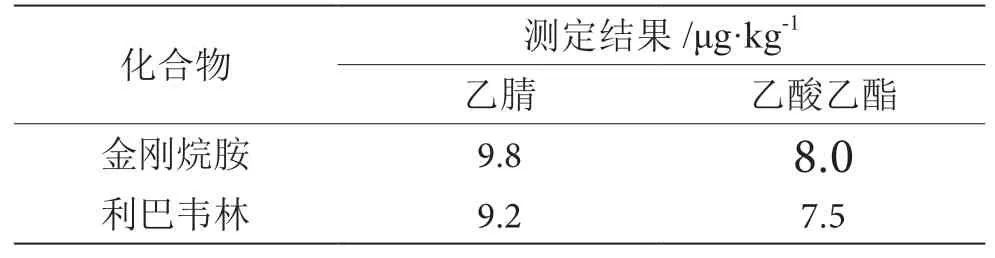
表1 空白基质测定结果表
2.2 酶解试剂选择
利巴韦林以原药、利巴韦林单/二/三磷酸酯形式存在,须对以磷酸酯形式存在的利巴韦林进行酶解,才能对试样中利巴韦林总量进行测定,采用37 ℃酸性磷酸酯酶对样品进行酶解,对比了非酶解、酶解2 h和18 h后利巴韦林总量的测试结果。结果表明,非酶解测得利巴韦林含量只有酶解2 h含量的2%,酶解2 h和酶解18 h后利巴韦林含量偏差在5%以内,因此选择37 ℃酶解2 h条件。
2.3 固相萃取柱选择
动物源性食品中兽药残留采用HLB固相萃取小柱进行净化,考虑固相萃取柱抗干扰、富集目标物能力等因素,本方法研究对比了HLB、C18、PBA 3款固相萃取柱,发现HLB对利巴韦林和金刚烷胺富集重复性和稳定性较差,C18固相萃取柱对利巴韦林和金刚烷胺富集能力低,平均回收率在20%以下,PBA固相萃取柱对利巴韦林和金刚烷胺两种化合物选择富集能力强,平均回收率能达到85%以上。因此本方法选用PBA固相萃取小柱对样品进行净化。
2.4 流动相选择
液相色谱串联质谱正离子模式对目标物利巴韦林和金刚烷胺进行检测,流动相中加入甲酸能够提高利巴韦林和金刚烷胺信号响应强度,加入乙酸铵能够改变利巴韦林和金刚烷胺峰形,本方法采用5 mmol·L-1乙酸铵水+0.1%甲酸作为流动相,梯度洗脱。液相色谱梯度洗脱条件见表2。

表2 液相色谱梯度洗脱条件表
2.5 内标定量和外标定量比较
采用外标法和内标法两种方式对样品进行检测比对,测试结果见表3,结果表明采用外标法回收率偏低,不满足检测要求,本方法采用内标法进行检测。
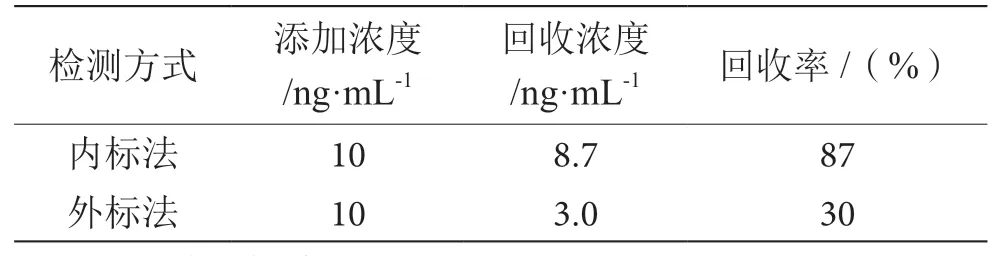
表3 内标法和外标法定量结果比对表
2.6 质谱特征离子选择
金刚烷胺、利巴韦林及其内标物在电喷雾电离源模式下采用SCAN全扫描,根据全扫质谱图中响应值高丰度好的离子作为母离子,进行产物离子扫描模式,将其母离子打碎以获得子离子碎片,选择响应高的碎片离子作为子离子。通过改变锥孔电压和碰撞能量优化仪器最强响应。金刚烷胺、利巴韦林以及其内标物的质谱条件见表4。
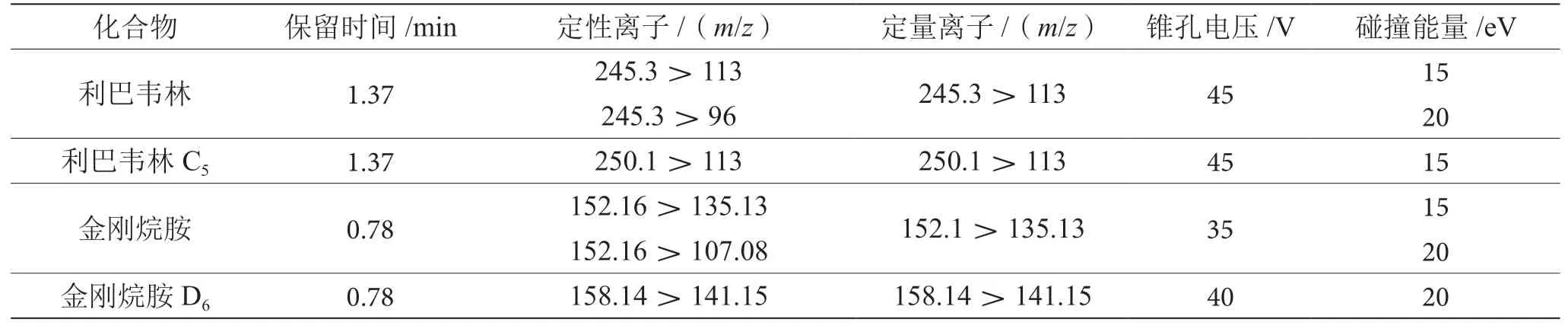
表4 保留时间、定性定量离子、锥孔电压、碰撞能量参数表
2.7 方法回归方法、线性范围及检出限
取浓度为0.5 μg·kg-1、1 μg·kg-1、2 μg·kg-1、5 μg·kg-1、10 μg·kg-1和20 μg·kg-1金刚烷胺、利巴韦林和浓度为5 μg·kg-1内标物配制标准工作液,液相色谱联用质谱进行测试,以外标物峰面积和内标物峰面积比值作为纵坐标(y),外标物浓度作为横坐标(x),绘制工作曲线,空白基质中添加两种化合物标准品,经检测,以3倍信噪比为检出限,以10倍信噪比为定量限,回归方程,线性范围、相关系数、检出限及定量限见表5。

表5 金刚烷胺、利巴韦林回归方程、线性范围、相关系数、检出限及定量限表
在0.5~20 μg·kg-1浓度范围内,外标物与内标物峰面积比值和外标物浓度呈良好线性关系,相关系数R2>0.99,检出限为1 μg·kg-1,定量限为2 μg·kg-1,满足分析要求。
2.8 精密度和加标回收
空白样品加标,经前处理提取、酶解、净化后进行测定,结果见表6。利巴韦林、金刚烷胺的回收率为85%~104%,相对标准偏差在0.8%~2.4%,表明该方法具有较好重复性及准确性,符合动物源性食品中兽药残留分析要求。
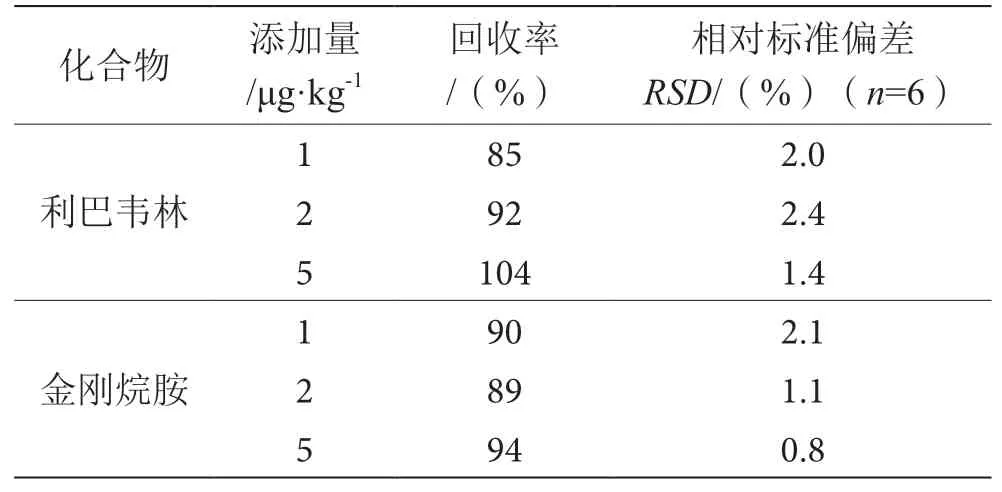
表6 加标回收率及相对标准偏差表
3 结论
本文研究了液相色谱串联质谱联法同时测定动物源性食品中利巴韦林和金刚烷胺的分析方法,通过提取、酶解、净化3个步骤进行定量检测,方法线性范围、检出限、定量限、添加回收率及相对标准偏差均能满足分析要求。方法操作简单,准确度和精确度高,缩减了利巴韦林和金刚烷胺检测时间,提高了效率,可用于动物源性食品中利巴韦林和金刚烷胺同时检测,并为其他食品中利巴韦林和金刚烷胺药物残留检测提供依据。
猜你喜欢
中国康复(2022年8期)2022-08-31
中国渔业质量与标准(2022年3期)2022-07-23
中国康复(2022年11期)2022-03-02
中国中医急症(2019年10期)2019-05-21
家庭医学(2018年8期)2018-10-17
中国卫生标准管理(2015年16期)2016-01-20
中国继续医学教育(2015年6期)2016-01-07
川北医学院学报(2015年5期)2015-12-05
中国医疗美容(2015年1期)2015-07-12
药学与临床研究(2015年4期)2015-06-05
