
15例Wiskott-Aldrich综合征患儿的临床特点及基因突变分析
2021-11-15 02:56郑湧智
福建医科大学学报 2021年3期
郑湧智, 李 健
Wiskott-Aldrich综合征(Wiskott-Aldrich syndrome, WAS),又称为湿疹、血小板减少伴免疫缺陷综合征,是一种罕见的严重的X-连锁隐性遗传性疾病,在存活男性新生儿中的估计发病率为4/106,临床上以血小板减少伴体积减小、湿疹、免疫缺陷、易患自身免疫性疾病和恶性肿瘤为特征[1-2]。WAS的致病基因定位于X染色体短臂着丝粒Xp11.22-p11.23,其编码的WAS蛋白(WAS protein, WASp)是肌动蛋白的关键调节因子,在非红系造血细胞中普遍表达,主要参与调节细胞分化、迁移、膜转运和细胞间通讯,还参与吞噬作用、细胞毒颗粒释放、抗原受体信号传递等多种细胞免疫功能[3]。WAS患儿异质性大,但基于WAS的基因突变类型及WASp的缺乏程度不同,可较好地判断患者病情的严重程度和预后,因此,熟悉二者的相关性对WAS的临床诊治至关重要[4]。鉴于WAS的低发病率,国内针对WAS患儿基因突变类型与其临床特征相关性的报道较少。本研究回顾性分析15例WAS患儿的临床资料,探讨WAS患儿的基因突变类型与临床特征的相关性。
1 对象与方法
1.1 对象 收集2014年1月-2020年6月诊治的WAS患儿15例,均为男性,发病中位年龄40 d(1~365 d),诊断中位年龄12个月(2~132个月)。1例有可疑家族史(舅舅自幼血小板减少,未确诊),余14例无家族史。WAS诊断标准参照文献[5],符合前7条中的1条或1条以上,且符合第8条:(1)男性,自幼起病;(2)具有血便、皮肤瘀点或瘀斑等出血表现;(3)反复皮肤湿疹;(4)反复或严重感染(以消化道、呼吸道及外耳道多见);(5)伴或不伴自身免疫疾病和恶性肿瘤;(6)血小板减少,伴平均血小板体积(mean platelet volume, MPV)缩小;(7)伴或不伴家族史;(8)WAS基因突变或WASp表达异常。依据WAS评分及分型标准进行分型诊断[4]:间歇性X-连锁血小板减少症(intermittent X-linked thrombocytopenia,IXLT)、X-连锁血小板减少症(X-linked thrombocytopenia,XLT)、X-连锁粒细胞减少症(X-linked neutropenia,XLN)以及典型WAS。
1.2 WAS基因突变检测 经患儿监护人知情同意,抽取患儿及其父母的外周血2~3 mL(EDTA抗凝)行基因测序(康圣环球集团北京海思特临床检验所)。提取基因组DNA,采用聚合酶链式反应(polymerase chain reaction,PCR)和测序的方法检测标本中WAS基因编码区的12个外显子,涵盖了该范围内的点突变、插入和缺失型突变,共进行12个PCR扩增反应和24个基因序列测定反应,具体方法见文献[6]。WAS基因编码含5个主要功能区的WASp,从N端到C端依次为Ena/VASP同源区1 (Ena/VASP homology 1, EVH1)或WASp同源区l(WASP homology 1, WH1)、碱性区(basic region,BR)、三磷酸鸟苷酶结合区(GTPase binding domain,GBD)、脯氨酸富集区(proline-rich region,PPPP)及C端的VCA区。
1.3 治疗及随访 患儿均在门诊进行随访,随访日期截至2020年8月31日。1例(例5)放弃治疗后失访,余14例接受治疗并随访。WAS评分≥3分的患儿,均建议行造血干细胞移植(hematopoietic stem cell transplantation,HSCT);<3分的患儿,建议定期输注静脉注射免疫球蛋白(intravenous immunoglobulin,IVIG)治疗,若有HLA全相合供者,可考虑行HSCT。
1.4 统计学处理 采用SPSS 21.0软件进行统计分析,计量资料采用中位数以及范围表示,计数资料采用百分比(%)表示。两样本率的比较采用χ2检验或Fisher确切概率法,采用Kaplan-Meier方法分析患儿的总体生存率(overall survival, OS),并行Log-rank检验。
2 结 果
2.1 临床特点 15例WAS患儿的临床表现及实验室检查如表1所示。首诊主诉最常见皮肤瘀点、瘀斑(10/15,67%),其次为血丝便(3/15,20%),另有1例(例1)因新生儿肺炎、败血症住院发现血小板减少,1例(例13)因先天性心脏病手术术前发现血小板减少。15例均曾误诊为免疫性血小板减少症(immune thrombocytopenia,ITP),接受大剂量激素及IVIG治疗,但血小板均未恢复至正常水平。整个病程中,11例有湿疹病史,其中3例呈持续性,8例轻度;10例有反复感染表现,6例呈重度,4例可控。
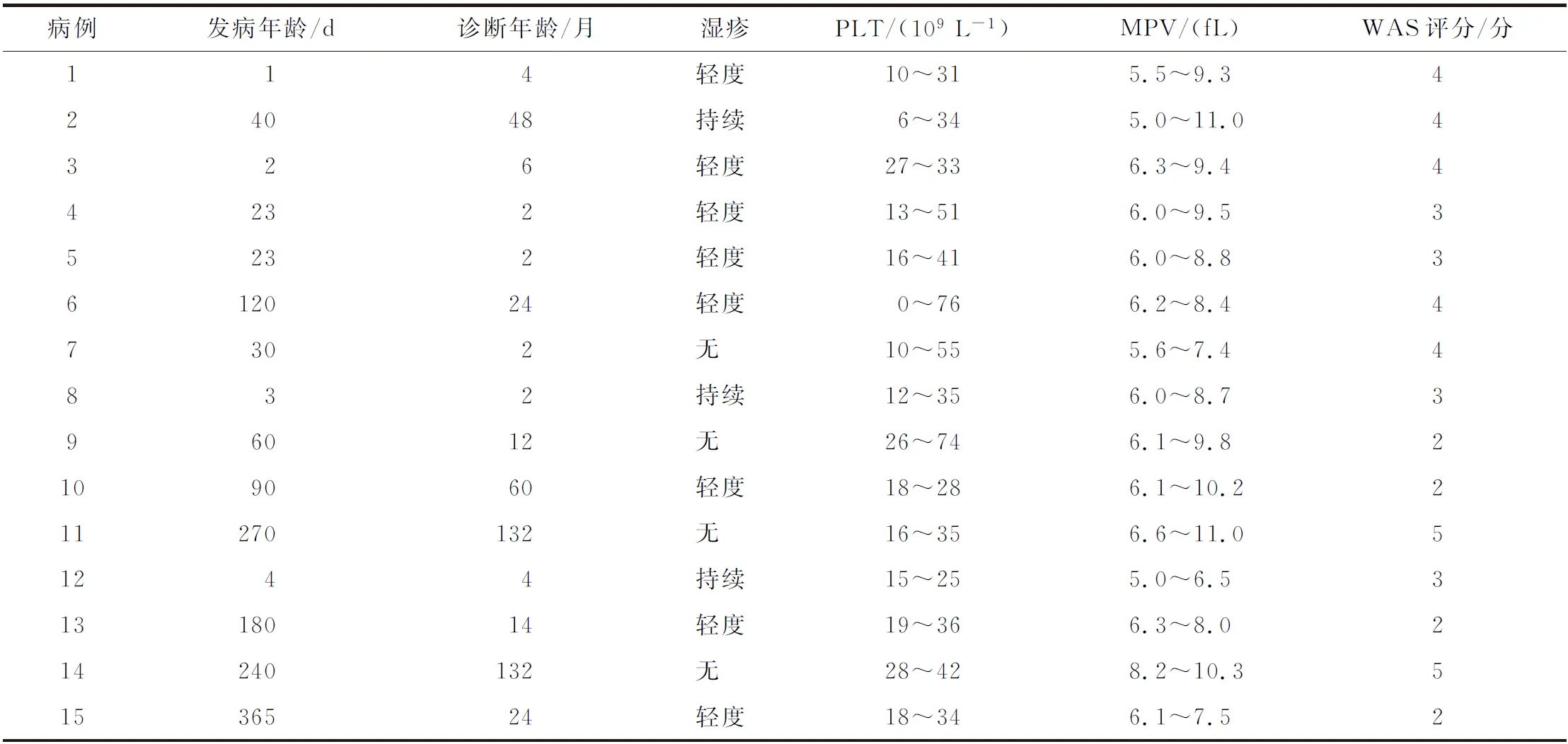
表1 15例Wiskott-Aldrich综合征患儿的临床特征及实验室检查
15例患儿均多次行血常规检查,血小板均减少,其中13例最低值<20×109L-1,13例MPV缩小。13例行骨髓常规检查,12例提示巨核细胞增多伴成熟障碍(其中9例伴缺铁倾向),1例提示巨核细胞减少伴缺铁倾向。15例患儿均行免疫球蛋白(immune globulin,Ig)和T细胞亚群(CD3+、CD4+和CD8+)检测,13例(87%)有Ig或T细胞亚群异常,其中IgG降低3例,IgM降低6例,IgA降低7例,IgE升高9例(中位值358 IU/mL);CD3+升高3例,降低1例;CD4+降低3例;CD8+升高4例,降低4例;CD4+/CD8+<1共7例。
WAS评分中位值为3分(2~5分)。例11于出生后9个月发现血小板减少,无反复湿疹、感染、便血病史。11岁时因颈部肿物首诊本院,行颈部肿物活检,病理提示:弥漫性大B细胞淋巴瘤,伴滤泡性淋巴瘤(3A级),EBER阳性;行WAS基因检测阳性,诊断为典型WAS(评分5分)。例13于出生后8个月发现血小板减少,无反复湿疹、感染、便血病史。11岁时因“发现皮疹、发热”首诊本院,诊断为系统性红斑狼疮,在治疗过程中反复发生重症感染(重症肺炎、血流感染),行WAS基因检测阳性(评分5分),诊断为WAS继发系统性红斑狼疮。
2.2WAS基因突变特点 15例患儿共发现14种15个突变,突变基因在外显子的位置及类型如表2所示。最常见的类型为错义突变(7例),均位于外显子1~4号,其中4例为XLT(WAS评分2分),3例为典型WAS(WAS评分3分2例,4分1例)。8例非错义突变位于外显子5~11号,表现为剪接突变、缺失突变、无义突变等,临床表型均为典型WAS(WAS评分4分4例,5分2例,3分2例)。典型WAS患者的比例,错义突变组显著低于非错义突变组(42.8%vs100%,P=0.026);WAS≥4分患者的比例,错义突变组也显著低于非错义突变组(14.3%vs75%,P=0.041)。

表2 15例Wiskott-Aldrich综合征患儿WAS基因突变检测结果
2.3 治疗及预后 中位随访时间15.4个月。1例(例5)于诊断6个月后失访;2例(例1、2)分别在4岁和4.5岁时行非亲缘全相合HSCT,移植后1 a血小板和免疫缺陷均得到纠正,目前仍无病存活。12例仅接受支持治疗:7例(例4、8、9、10、12、13、15)仍存活,血小板为15×109~72×109L-1;3例(例3、6、7)因反复重症感染死亡;1例(例11)继发弥漫大B细胞淋巴瘤,予2个疗程减低剂量的利妥昔单抗+CHOP方案化疗后,淋巴瘤达完全缓解,建议行HSCT,但家属因经济问题仍在考虑,目前随访2.9个月,继续巩固化疗中;1例(例14)继发系统性红斑狼疮因合并反复感染后才通过基因检测诊断WAS,予泼尼松和羟氯喹治疗,出现肺部感染、血流感染、消化道出血、睾丸坏死等并发症,且系统性红斑狼疮不能控制,出现狼疮性肺炎,予大剂量甲泼尼龙琥珀酸钠(甲强龙)冲击、吗替麦考酚酯等治疗,病情仍无法控制,家属放弃治疗,数日后死亡。仅接受支持治疗的12例患儿中,按基因突变类型不同,错义突变组2 a总体生存率高于非错义突变组,但差别无统计学意义(100%vs20.8%,P=0.06,图1A);按WAS评分不同,WAS评分≥4分的患儿2 a总体生存率显著低于WAS评分≤3分的患儿(26.7%vs100%,P=0.008,图1B)。

A:不同基因突变类型患儿的生存率;B:不同WAS评分患儿的生存率。
3 讨 论
WAS异质性大,临床表现多样,其中IXLT或XLT仅表现为血小板减少,易被误诊为ITP;XLN仅表现为中性粒细胞减少,也不易诊断。即使是典型WAS,也多在反复感染后才得以确诊。我国WAS病例的系列报道显示,WAS的发病年龄为15~90 d,诊断往往滞后,易误诊为ITP等疾病,确诊年龄为10~24个月[7-10]。误诊和漏诊可导致WAS患儿反复感染,并进展为自身免疫性疾病或恶性肿瘤,导致治疗难度增加和复杂化,严重影响患儿的生活质量,甚至危及生命[4]。因此,熟悉WAS的临床特征,有利于避免漏诊并早期诊断WAS。
本组15例,发病中位年龄40 d,确诊的中位年龄12个月,均曾误诊为ITP,其中1例患儿(例13)于出生后8个月发病,诊断为系统性红斑狼疮,因反复重症感染于11岁时行基因检测才得以确诊,但因此时治疗难度大,最终死亡。可见早期诊断非常必要。详细的病史对WAS的早期诊断至关重要,对于发病年龄小且免疫治疗效果不佳者,需高度怀疑。若多次血常规检查提示MPV缩小、湿疹、反复感染病史,则强烈提示WAS可能,应进一步行基因分析。另外,目前多数基因检测主要针对WAS外显子,对于有WAS典型表现但外显子基因筛查阴性者,需进行全基因组测序以排除内含子区域的剪接位点突变。另外,由于X染色体可能随机失活,也有女性患病的报道,对于有典型WAS临床表现的女性患者,也不能排除本病,应进行基因蛋白分析。
国内报道多为典型WAS病例,而XLT、IXLT、XLN等则少有报道,其中WAS评分5、4、3和2分患者的比例分别为27.8%、27.8%、38.9%和5.5%,可见重症(4~5分)患者的比例>50%[4,7,9-12]。这可能与轻症患者易漏诊、误诊以及报道偏倚有关。本组病例中,典型病例占73.3%,与报道基本相似[7, 9]。
WAS基因由12个外显子组成,编码502个氨基酸组成的WASp,而WAS基因突变可导致WASp表达量下降或完全缺如,WASp的功能也相应部分或完全缺失[13]。目前已报道400多个WAS基因突变,最常见的是错义突变,其次是剪接、缺失、无义及插入突变[14]。大多数错义突变位于第1~4外显子,剪接突变则常发生于第6~10内含子,缺失、无义及插入突变则散在分布于WAS基因的各个部分[15]。本组病例所报道的突变类型及位置均与报道基本相符[7-9,16]。
突变类型和突变位置与WAS的临床表型及预后相关。散在分布WAS基因上的无义、插入、缺失或复合突变,导致WAS蛋白完全不表达或表达截短型蛋白,多表现为典型WAS;WAS基于第1~4外显子的错义突变,导致WASp表达不完全,表现为XLT[16-17]。另外,WAS突变类型与WASp活性有关,后者有助于判断临床严重程度和预后,但因笔者科室未开展该项检测,故未能进行相关分析。本组病例中,错义突变组典型WAS患者及WAS评分≥4分患者的比例均显著低于非错义突变组,说明基于突变类型与临床表型具有明显的相关性;错义突变组2 a总体生存率高于非错义突变组,但差别无统计学意义,表明突变类型可能与预后具有相关性,有待扩充样本量进一步明确。
WAS的临床异质性大,治疗方案需个体化。对于XLT患者,应用支持治疗可能获得较好的预后[7]。对于典型WAS患者,HSCT是目前唯一有效的根治手段,且远期预后良好[18-19]。文献报道,接受HSCT的WAS患儿总体生存率>90%[1,7,9,11,20]。近年来,先通过基因编辑技术对患者造血干细胞的缺陷基因进行修饰,再行自体移植,可消除同种异体反应的风险,从而获得较好的临床改善[21];对于缺乏合适供者、有严重并发症等情况的典型WAS患者,使用基因修饰的自体干细胞移植或将成为治疗WAS的标准疗法[22]。如何把握移植的时机?研究显示,随着年龄的增长,继发自身免疫性疾病或恶性肿瘤的发病率逐渐升高[4],导致治疗难度增加和复杂化,最终可能失去移植的机会。一项国际多中心研究显示,在129例接受HSCT治疗的WAS患者中,<5岁组的总体生存率显著高于≥5岁组(94%vs66%,P=0.000 8)[20]。本组病例中,WAS评分≥4分的患儿中,2例接受HSCT均长期生存,3例未能接受HSCT的患儿因反复感染死亡,另外2例在疾病早期仅表现为XLT,但1例在11岁时继发系统性红斑狼疮,1例在11岁时继发淋巴瘤。因此,对于重症WAS患者需尽早行HSCT,而对于非典型WAS,由于疾病状态无法自行逆转,其病情不断进展,也应长期随访和动态进行严重程度评分,及时进行HSCT,预后更佳。
总之,WAS患儿发病年龄小,临床表现缺乏特异性,易误诊;详细的病史采集,及时的基因检测有助于早期诊断。WAS基因突变类型与临床表型及预后有一定的相关性;长期随访,进行动态WAS评分,有利于制定合理的治疗方案。
猜你喜欢
分子诊断与治疗杂志(2022年9期)2022-10-09
祝您健康·文摘版(2021年7期)2021-07-17
祝您健康·文摘版(2020年7期)2020-07-13
世界知识(2018年19期)2018-11-21
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
世界知识(2017年5期)2017-03-18
湖北农业科学(2014年11期)2014-09-10
